
ATYR Follow-up
Final look before topline

Disclaimer: This report is a continuation of the previous report published July 29th, 2025. While the authors believe that the clinical trial will fail to meet its primary endpoint it is still possible that our analysis and conclusions are wrong. In the event that the topline results stat sig the stock could trade 200-500% higher on open. Shorting the stock into the readout, represents extreme risk and we discourage such risk taking behavior. We do not give financial advice and we do not endorse shorting the stock given the asymmetric risk- this is an educational write up.

Fourier Transform Research
____________________________________________________________________________
Over the past few weeks we have received some criticism of the previous report. Some of these rebuttals meaningfully engage with the substance of our arguments and some fail to directly address the claims or arguments made previously . As fans of healthy and open debate we have compiled some of these comments, and our replies to them below.
Our original report argued:
Many patients won’t need long term steroids due to milder disease state
This is a good point and we have prepared a rebuttal below. We have also found clinical data to anchor a relapse rate analysis to, and we predict the placebo arm performance in the ph3 trial.
A clear baseline imbalance between placebo and higher dose cohorts
The rebuttals we’ve heard do not meaningfully engage with our arguments and so they still stand. We think that the steelman on the bull side (as some have said) would be to argue that there is an imbalance, and the drug still works beyond that imbalance. We still disagree, but we have added a bit more color to that as well.
A graveyard of failed MoAs using adjacent pathways to the NRP2 MoA in sarcoid
The MoA arguments were never refuted. Asserting that this time is different does not refute the problems with the MoA we highlighted in the first report. However, for fun, we have gone into more detail on this MoA using computational simulations, as well as examining the efzo functional data, binding data, epitopes, and doing a detailed breakdown of the MoA biology.
Low level of evidence for pre-clinical efficacy translating to successful in vivo efficacy
This point stands, and we have added more color to it.
Disclosures: This Report is provided by Fourier Transform Research solely for educational purposes and constitutes impersonal, generic commentary based entirely on publicly available data. The views expressed herein reflect the personal opinions of the author as of the date of publication; they are not tailored to any individual’s financial situation or objectives and do not constitute investment advice, a recommendation, or an offer to buy or sell any security. The author holds a short position in the securities discussed. No part of the author’s compensation is, was, or will be directly or indirectly related to any specific recommendation or view contained herein. The author has not received—and will not receive—any payment of any kind from the company(ies) whose securities are covered by this Report. This Report contains no material non‑public information. Trading the securities covered herein is subject to a blackout period for the author commencing 72 hours before and ending 24 hours after the Report’s public release. All factual data are believed to be accurate at the time of writing, but Fourier Transform Research makes no warranty as to its completeness or accuracy; recipients should conduct their own due diligence. Past performance is not indicative of future results.
This patient population is sicker than you think, there won’t be spontaneous remitters
This is an important argument to clarify because some have read our report and believe that a lack of spontaneous remissions would imply that everyone relapses quickly. If people aren’t spontaneously remitting after a taper to zero in the ph3 clinical cohort then the drug must be doing something since we know there is separation. To address this claim we’ve been engaging with bulls about why they believe that this cohort is in fact unlikely to remit and likely to relapse to OCS. The steelman here is that they are patients with documented, severe disease, confirmed by biopsy, and they are on steroids when they join the study.
Those facts don’t necessarily tell us that these patients won’t remit on trial, though we appreciate that they do indicate the progression of disease. The guidelines tell us that patients who present in the clinic with relatively normal lung function are not usually prescribed OCS until further disease manifestation. This is because early steroid use (and toxicity) isn’t needed when patients remit so often, as noted in our last report.
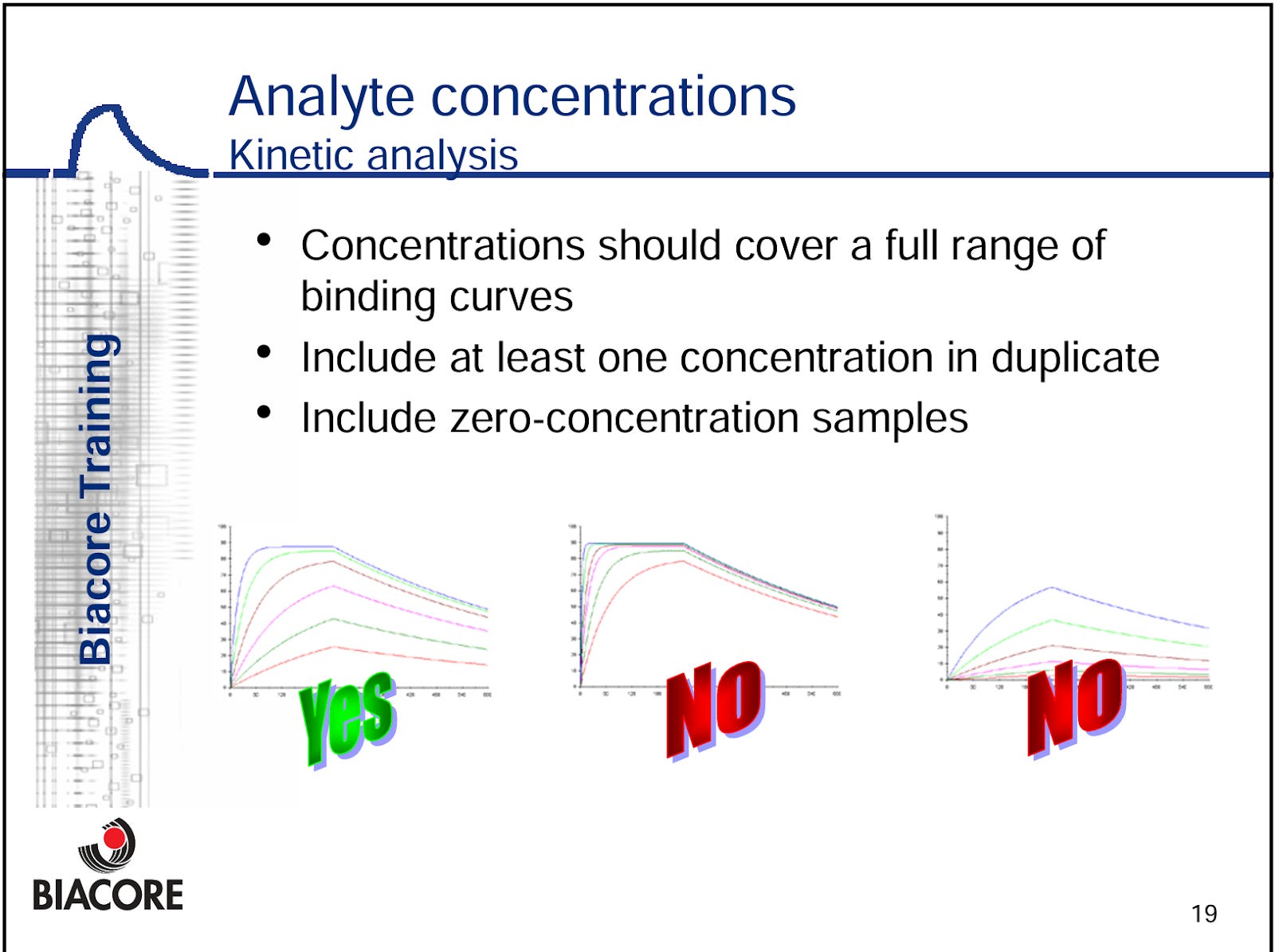
The only baseline data we have going into the ph3 readout are the MRC scores disclosed at ATS2025, which tell us that over half the patients are MRC score 1, and ~90% are 0-21. This is certainly more mild than the 65% of pts who were MRC 1-2 in the ph1/2 data set2. However beyond this data we have very little to go off of to assess disease severity. We will note that there are 101 (38.3%) of patients who are taking a concomitant immunosuppressant. Some might say this is directly related to the disease severity. We think that’s fair to suggest, but not necessarily proven. We believe some clinicians are using methotrexate prior to disease progression as a steroid sparing agent in earlier-line settings, as some patients can’t handle steroid toxicity or have significant QOL concerns. Further, given the non-inferiority noted in the highly anticipated PREDMETH study recently published in the NEJM this year, we think clinician experience is bolstered this study3.

Beyond this, clinicians we’ve spoken to have said that in their practice they aim to use MTX earlier. However its use as a front-line agent is not reflected in the guidelines, and the PREDMETH study was only recently completed and published so we will only share our anecdotes as such. We believe some centers reserve MTX use for severe cases, and some use it in front-line settings.
All that said, we note that there are no great baseline variables that conclusively stage this disease. After an initial course of treatment what are the odds of relapse?
This can be a very difficult question to answer since patients can have varied outcomes based on where they start in the course of their disease, and precisely how much treatment they receive. We sought to dig further into the literature on this point because it is our contention that patients have been over treated with steroids for many years.
Gottlieb tried to address this question in 1997 with his study on outcomes in sarcoidosis, where he enrolled 337pts over 4yrs and had a spontaneous remission group, an induced remission group who took OCS and discontinued, and a recalcitrant group who could not last more than 1mth without OCS. We note that these were patients with a long course of disease (~10yrs) and 73% of the induced group were African-american, which the author notes relapse more often than their Caucasian peers. They also note that in this study most relapses happened within 2-6mths after discontinuation of steroids, but late relapse was also not unusual, happening in over 20% of such patients4.

We can see that, commensurate with current guidelines, a longer course (12-24mths+) of OCS therapy is unlikely to lead to remission of disease. We note that roughly 9/18 of pts treated for 2yrs or less relapsed after 1yr in this study.
Baughman in 2006 tried to continue this effort in his ACCESS study which enrolled over 200pts with 2+yrs of follow-up data who had confirmed sarcoidosis by biopsy and who were treated within 6mths of that diagnosis. Patients were evaluated for treatment at baseline (B), during an interim follow-up (I), or at a later follow-up (F), 18-24mths after the initial visit. Many of these patients (72%) were stage I/II by chest x-ray, and we also note that 45% were African American5.


And here we see that those early-stage patients who haven’t lost much lung function fared quite well and many never needed treatment in the 2yrs of study collection. However we do see that many patients in this cohort who had progressive disease were taking OCS over the course of the study. In a different publication on the same data set, they also noted that of the 110pts who were on OCS at baseline 53% of them were able to discontinue therapy. We also note a substantial number of patients with level 1 dyspnea scores remitting spontaneously- we count 15/69 (21.7%).
Further, we see Bilgin in 2023 try to gain insights into sarcoid treatment as a function of physiological markers of disease6. They took baselines in every conceivable metric including spirometry (FVC, FEV, DLCO), age, length of disease, length of treatment, biomarkers…etc. and conclude that these are all valuable to grouping patients but we don’t have a clear recommendation saying that if patient hits X value on Y test treat with this specific amount of Z drug.
It’s good to go into the depths of the literature and find out how we came up with the current guidelines but obviously over time things change. Recently published paper by Partin et al. show us that there is still no consensus on how to treat sarcoid with steroids7.


We know that the suggested guideline is to treat with steroids for a year while tapering, but this practice has low level of evidence supporting it. It appears to us that it is driven by clinical practice which is nearly half a century old8,9. Coming back to whether to use OCS or MTX, while the current recommendation is to use OCS over MTX, this is not supported by evidence and is anecdotally driven, as per the 2021 ERS guidelines10.
Where does that leave us on the topic of this ph3 clinical trial?
We know that there is no established, rigorously determined guideline for how to treat front-line sarcoid patients
This ph3 trial should help us establish a new guideline whether efzo is a new drug or not
Let’s take a step back and take apart the endpoint one last time. There has been a lot of debate about the semantics of our argument. Bulls will say that of course patients will relapse. They have taken our argument to say something like: patients will spontaneously remit, and never need drug again.
This is not what we are saying, and this is also not the question the ph3 study will answer.
The ph3 primary endpoint is: the amount of OCS patients are taking, per day, between weeks 45-48.
This is very important to understand because we’re not saying that placebo arm patients will never relapse, we’re saying that between weeks 45-48 we think it is highly unlikely that there is a clinically meaningful or statistically significant reduction in daily OCS usage between drug and placebo arms. We think this large scale, randomized placebo-controlled trial will ultimately reshape the guidelines towards a steroid sparing paradigm.
This study is not testing whether there is even a reduction in steroids over the course of a year, it’s specifically testing whether we see a reduction in steroids usage between weeks 45-48. This is very important to understand. We don’t have to contend with whether patients on this trial will ever relapse, we think some will, but the question is whether they will relapse and be on OCS between weeks 45-48.
Let’s also keep in mind a few additional points about this trial:
Patients can receive rescue taper if they can’t sustain the initial taper, up to 3 times
Almost 40% of patients are taking MTX, and are permitted to continue taking MTX throughout the entire study
Our contention is that most patients benefit from the use of OCS quite quickly and need to taper off sooner than the current clinical guidelines suggest. We reference the above PREDMETH study showing much of the functional benefit is observed by wk 163. Further, we see that in a recent Dutch study using at-home spirometry analyzers patients derive most of their benefits within the first month- improving FVC, DLCO, and lowering dyspnea scores11. This trend holds up quite well in the patients with better baselines but again we note (as we did in the first report) that the lower baselines confer a poorer outcome in most patients.

Further, we have found the very useful 86 patient SARCORT study specifically asking what is the rate of relapse in patients who taper to 0mg/d of steroids over 6 months after an induction regimen of 20mg or 40mg. We see that baselines are quite comparable to the EFZO-FIT ph3 study (56% g0-1, 44% g2+), as these are patients with active disease and who are unlikely to spontaneously remit, hence treatment with OCS12. This study provides us with a very useful kaplan meier curve to assess relapse rates in these patients receiving OCS treatment.



We note that patients are plotted from the point of randomization, which was when they started treatment, and they are on drug to taper until 6m. We have remade this KM curve, omitted these early relapses because early relapse patients will be permitted a rescue tapering up to 3 times. If they can’t taper after 3 tries they will continue on trial with OCS treatment. In this way patients could be finishing their taper much later in the study before the final wk45-48 readout.
Next, we referenced Fang et al.’s paper which showed that patients who used a full course of MTX (permitted on EFZO-FIT) were able to remit the disease and only relapsed ~18% of the time in their study, with a mean follow-up of 13mths13. We also found a front-line MTX vs steroid trial in which the patients who completed treatment had an 8.3% relapse rate at 12m, but we will only rely on the Fang et al. paper to be conservative14. These data can help us handicap the OCS discontinuation study and estimate the probability of relapse at wk45 for the EFZO-FIT placebo arm, since 40% of them will have MTX.

This estimate suggests that the placebo arm in the EFZO-FIT trial will have a relapse rate of 25% at wk45.
If we go further and assume that 75% of pts are therefore at 0mg/d of prednisone, 20% at 10mg/d, and 5% at 15mg/d, we see a net reduction in steroids to 2.75mg/d (!) in the placebo cohort.
We believe this result alone would be practice changing. Indeed, if patients are able to go to 0 OCS after a 3mth taper schedule then this would suggest that patients have been chronically overtreated with OCS for sarcoidosis for many years. However, we think it’s important to point out that this does not mean that a substantial number of patients will not relapse at month 18 or 24, as the root of the disease is fundamentally not being treated. That said, saving patients from the toxicity of chronic steroid usage alone will lead to substantive patient benefit.
As we will show below, we think the drug is inert against sarcoid. In our view it’s possible the drug has a small effect, but it’s also possible the drug is worsening the disease for patients as well. We predict there to be no effect one way or the other, and placebo will be about as effective as the drug, and efzo will fail to hit stat sig. To put a number on it we predict the placebo adjusted delta between wks45-48 for efzo 5mpk to be <0.5mg/d, although we wouldn’t be surprised if placebo takes another round against drug developers.
Baseline Imbalances
As we discussed extensively in the first report, we believe there was a significant baseline imbalance that exaggerated the positive results of the ph1/2. All of that argumentation stands, unrefuted, save for a comment about the baseline OCS to final OCS dose being adjusted for baseline via an ANCOVA. That argument does not engage with patient specific baseline scoring, so our arguments stand.
Some have questioned our use of a chat GPT generated patient level data set, and that the biomarker analysis was “difficult to interpret”. This is not refutation and does not engage with the substance of the argument and therefore our original arguments stand. There was in fact a large baseline difference in the patient level biomarker data, and our simulated patient data completely recapitulates the available biomarker data from the ATS2022 poster.
Going deeper into the PK paper by Walker, we note that a lot of the “downside” so to speak in the placebo group is seen by a couple of patients, with placebo otherwise performing quite well compared to the rest of the cohorts15. We also want to highlight the one patient on the far right of the dose range as they had some difficulty clearing the drug and therefore had longer dose exposure than any other patient. Despite this much longer exposure, they did not perform the best in the cohort (discrediting the dose dependent response), nor did they outperform the best placebo patient in OCS % reduction or FVC improvement.
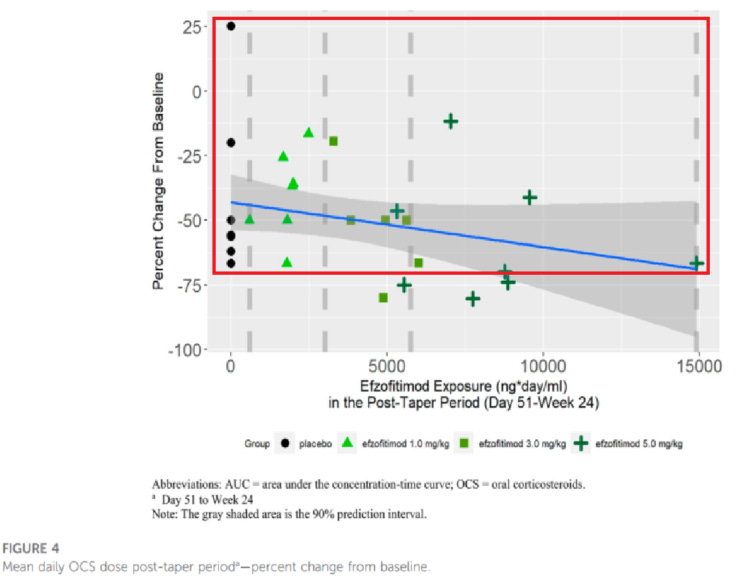
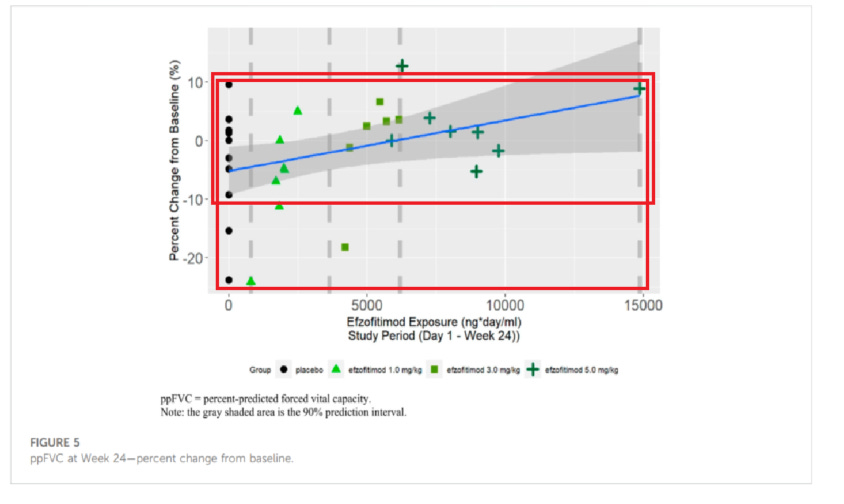
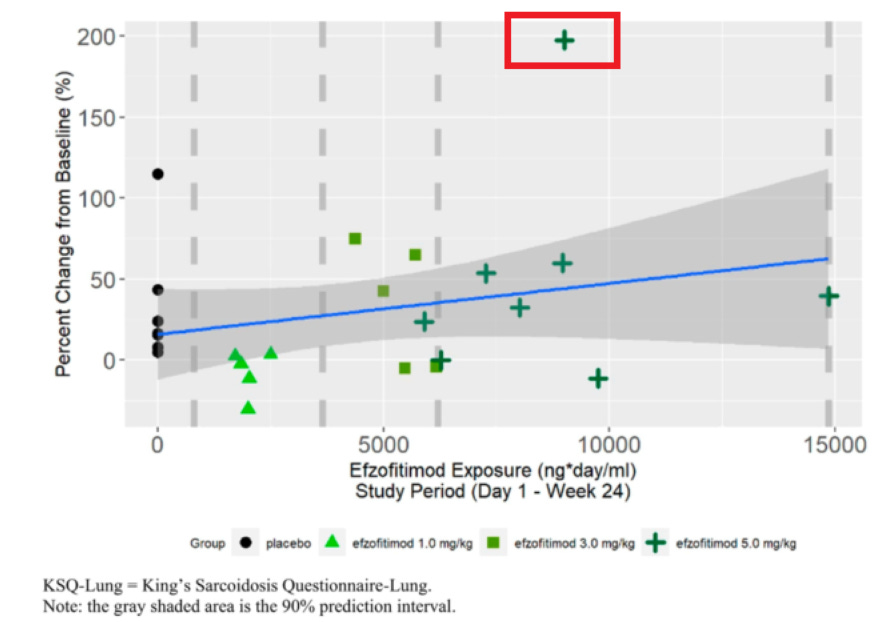
We think this data offers more evidence of a baseline imbalance than a dose dependent response, since we don’t see outsized improvement to FVC in drug groups (particularly the overexposed patient) and the downside in pbo arm appears to be driven by outliers. The KSQ score benefit for the 5mg/kg group in particular is clearly dominated by a single outlier patient, and we note from the Chest paper pre-print that this so called benefit was observed after the first month of reporting. So if efzo works, it works very quickly in that dimension, however it could also be the case that patients getting off steroids feel better as long as disease doesn’t re-appear.
We also want to re-examine the clinical trial design in the ph1/2. It was a multiple ascending dose (MAD) study, which enrolled patients 2:1 drug:placebo in each arm which ended up being 8:4 across all cohorts. They started with the 1mg/kg group, then 3mg/kg and finished with 5mg/kg. This is important to remember since they ran patients in groups and both clinicians and patients were unblinded to their cohort. They didn’t know whether they got placebo or drug, but if a patient was in the 5mg/kg group it’s likely they were biased towards a positive patient reported outcome via the KSQ score. We also note that through the trial they changed protocol a couple of times.
In particular, we note the removal of a 90% ppFVC as a ceiling for eligibility. This change was made fairly early in the study but surely some of the 1mg/kg cohort had already been enrolled prior to this date.


We also note that they removed the requirement for PET/CT in 2021, and therefore couldn’t confirm a recent lung parenchymal activity thereafter. We acknowledge the limitations present during COVID but this likely benefitted later populations in the study compared to earlier lines.
In presentations CEO Sanjay Shukla also mentioned that there were more fibrotic disease patients enrolled in the 1mg/kg group, before this change was made. This difference in protocol particularly mid-study significantly confounds the randomization efforts, particularly in the placebo arm as one third of the pbo arm was randomized sequentially, which means that the 1mg/kg cohort contained these fibrotic lung patients whereas the 5mg/kg cohort did not. This clearly explains the poor performance of the 1mg/kg group particularly compared to the rest of the study groups.
We also note that the protocol only made the taper to 0mg/d OCS option available much later in the study, as per 2019 and 2020 10K filings for aTyr. This is important to mention because it means that not every cohort (and not every patient within any cohort) was given the chance to taper to 0mg/d. So when aTyr claims there were more patients who tapered to 0 in the 5mg/kg group compared to placebo, this is confounded by the fact that there were 8 patients in each drug arm given the chance to taper to 0 and only 4 in the placebo arm. This means the correct denominator to use in the placebo cohort tapering to zero is 4, not 12, as some have suggested.
It is inappropriate to compare the placebo adjusted results of the study, and we are troubled by the notion of comparing the so-called “sub-therapeutic” and “therapeutic” doses when the latter benefitted from a healthier patient cohort by definition due to protocol changes. No study is perfect but a post-hoc retrospective sub-grouping of cohorts without mentioning the protocol changes is problematic to say the least.
Mechanism of Action-CCR5 conflict untrue, no one replicated the Howard et al. work
In our first report, we suggested that it’s possible the active binding domain in efzofitimod, HARSWHEP2-60, may be trafficking CCR5 bearing lymphocytes to lung tissue as a constituent element of the HARSWHEP2-60 fragment, HARSWHEP1-48, is contained within16. Company management has stated that the paper supporting this assertion has never been replicated and that efzo is not in fact chemotactic for CCR5 bearing cells. Further, they state that their Retrogenix screen was monospecific for NRP2 and that the assay did not light up CCR5 which was included in the assay.
We would add that the Retrogenix assay typically fixes its cells to the wells of the microarray it screens samples on. Fixation of cells can in some instances lead to changing the conformation of some proteins. This can lead to false-negative readouts, as seen in the Norden paper17:
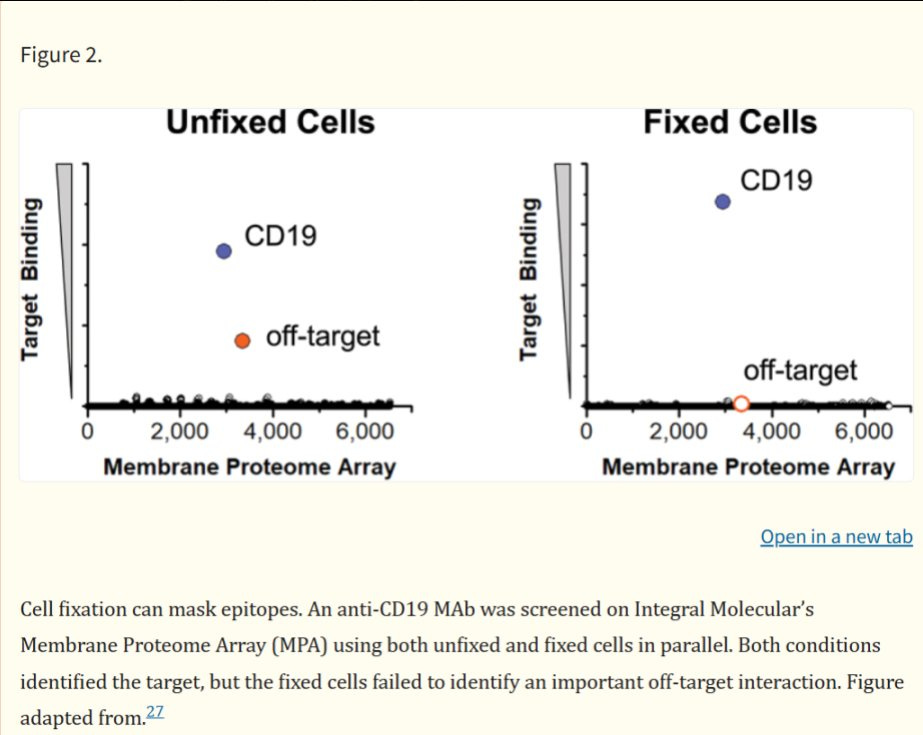
Thus, it is possible that CCR5 binding was abrogated due to the fixation of cells to the well, and we can’t rule out the possibility that HARSWHEP domain has a high affinity for CCR5.
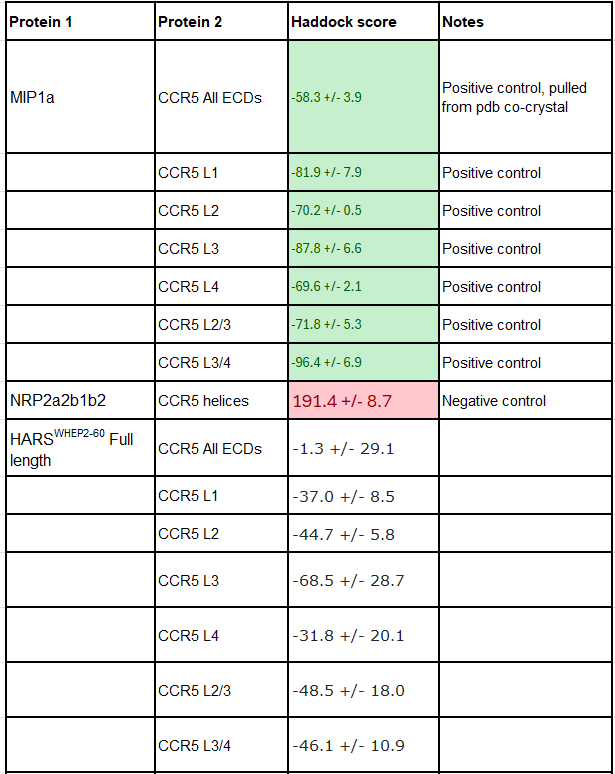
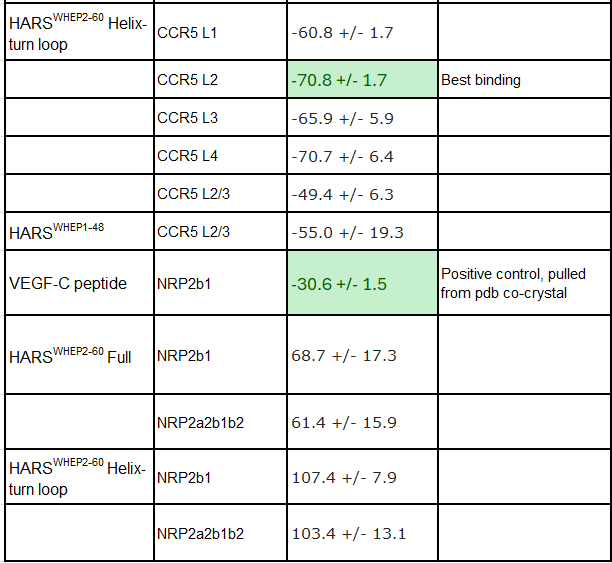
In order to address this issue as best we can, we decided to run computational simulations of various constructs using HADDOCKv2.4. We acknowledge the limitations of purely computational based docking experiments compared to lab based experiments, but we believe they serve us well in ranking the potential binding of the HARSWHEP domains and the proposed ligands NRP2 and CCR5.
First, let us be clear and say again that HARSWHEP1-48 is contained within HARSWHEP 2-60. The difference between the molecules is the inclusion of a c-terminal tail in the latter. Outside of this tail, they are the same molecule.
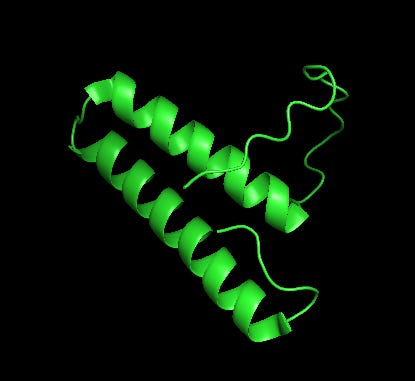
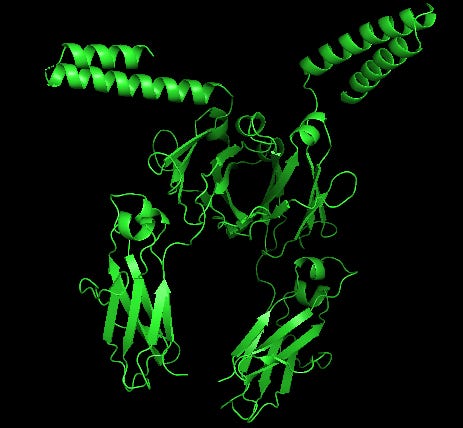
In our first paper we referenced the Howard et al paper which showed that HARSWHEP1-48 bound to CCR5 and was capable of acting as a chemotactic agent. They deduced that the likely epitope was ECL2/3, although we don’t know the paratope on HARSWHEP1-48. So we tested multiple different configurations of the HARSWHEP1-48 to CCR5 and found strong binding scores when using the full length construct as well as when using only the helix-turn-loop, as aTyr proposes efzo binds to NRP2.
We observed a lower docking score for the HARSWHEP1-59 domain as compared to the HARSWHEP 1-48 domain in these full-length simulations, though the helix-turn binding strength is conserved in both models.
That said, recapitulating binding of either HARSWHEP domain to NRP2 proved more challenging. We did successfully find some potential binding configurations using this approach though they scored much lower than CCR5. Both the full length and helix-turn loop configurations had low Haddock scores, implying they would have difficulty binding the target.
To be clear we would not suggest that this proves the molecule has no binding to NRP2, as we said earlier, but rather that this helps us map out the likelihood of whether HARSWHEP1-48 HARSWHEP2-60 ultimately bind NRP2 and/or CCR5. We still believe they both bind, and would add one more piece to this, taken from aTyr patent US8404242B2.
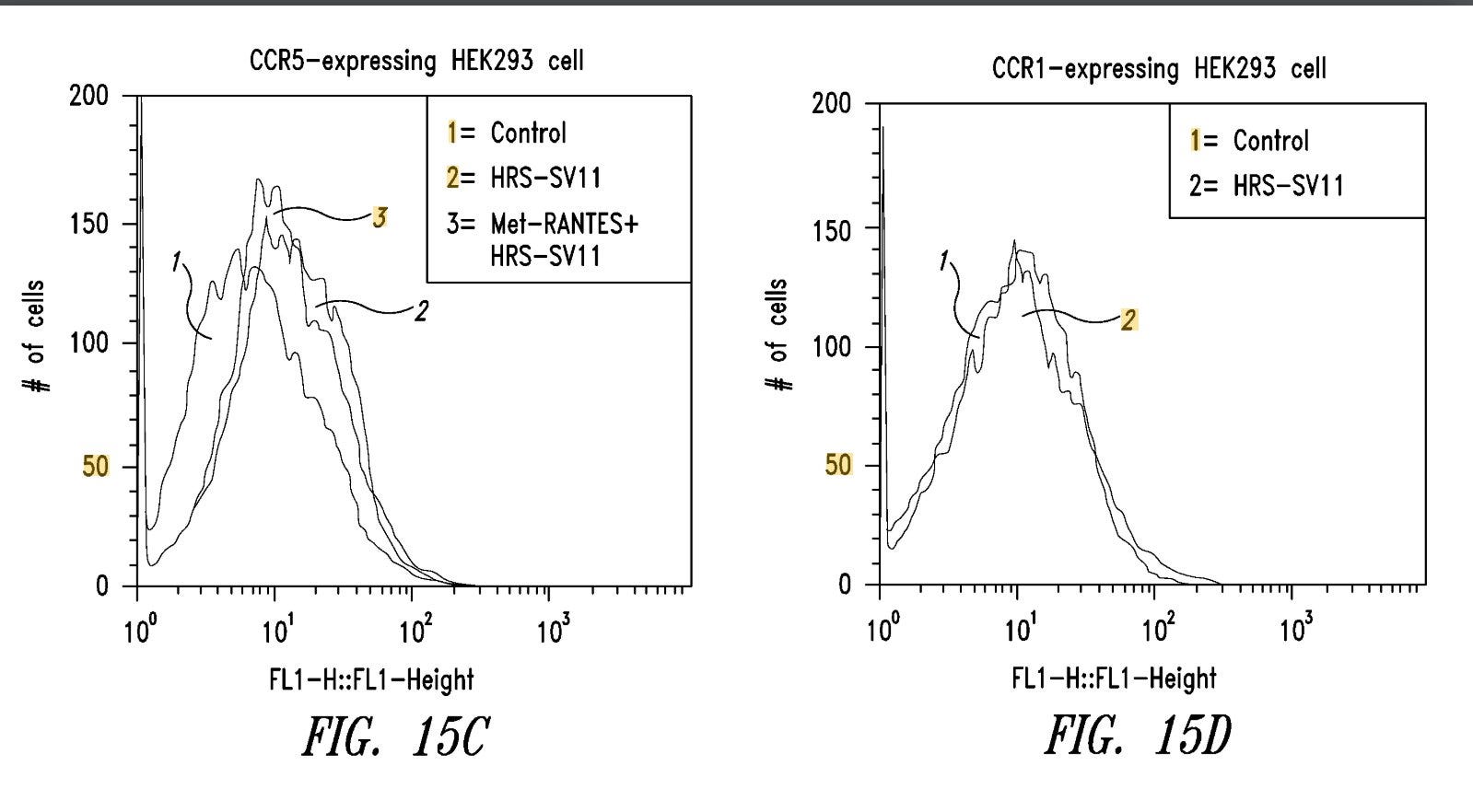
Where we see that a construct built from the WHEP domain binds extremely well to CCR5, shifting the fluorescence signal completely to the right. This is not the same HARSWHEP2-60 domain used in efzofitimod, it is a novel construct with a longer tail and a di-cysteine residue “hook”. But we find it interesting nonetheless that another HARSWHEP domain variant exists with the intention to bind and manipulate CCR5. Clearly, the HARSWHEP domain is a good backbone to manipulate CCR5 and even aTyr believes it since they patented the molecule.
That said, below is a table summarizing our computational findings. Negative scores are better, and you can read more about how they work on the Haddock website. We tested MIP1a against CCR5 extracellular domain loops sequentially and against all of them at once as a positive control, and then used the HARS WHEP domains- using the full length variants as well as the middle helix-turn loop structure. Similarly, we tested HARSWHEP against two different NRP2 domain structures with a positive control VEGF-C co-crystal peptide, and HARSWHEP. Lastly, using CCR5 alpha helices and NRP2 a2b1b2 domains we created a negative control against both proteins ensuring the model functioned correctly. All structures were pulled from pdb and are from crystal or co-crystal structures. Further details about each run and more images of binding events are given in the Binding Data Appendix.
EC50 does not equal affinity
Beyond these computational analyses, we struggle with the wet lab work done for efzo.The fact that aTyr has never reported a KD value for their lead candidate is troubling. They have never published this value in their Science paper or the supplemental material18. It has never been put on a poster. We can’t find it in the patents. We acknowledge the use of a single sensogram from an SPR assay at 150nM shown in the ATS2020 poster19, and this same graph used in their Science Translational paper, but one can not derive a KD value from a single sensogram and they only ever claim that efzo is a nanomolar binder- 1nM and 999nM are nanomolar binders. Without stating it explicitly, it is impossible for anyone to know where on this spectrum efzo lies. Target engagement is critical for a drug to have a physiological effect, and thus far the lack of KD invites more questions than it answers on that front.
The closest data piece we have is an EC50 value on HEK293 cells overexpressing the NRP2 target protein with a stated EC50 of 30nM. But this is a highly problematic assay to claim affinity for a target. EC50 is telling us when the assay is reporting half the full fluorescence value. In this case we can see that they are flowing efzo over cells overexpressing NRP2.

The trouble here is that we have to make sense of the cell data. Are there more copies of NRP2 on these cells than sarcoid patients cells?
In the supplementary figures S1E from their Science paper (left), we can see that the NRP2 WT HEK293 cells they used for EC50 calculations have high intensities, roughly 600k MFI on this device. On the right, we see their review paper which references the ATS2020 poster by Paz et al., hitting roughly 20k MFI18,20.


This gives us two bar graphs with very different intensities. We searched but can’t find the instrumentation used to generate the figure on the right (sadly), but we infer an order of magnitude delta in terms of the signal detected between these two sets of cells expressing NRP2. This implies that the copy number for NRP2 on the Science paper cells is much higher than the sarcoid patients in vivo cells. Thus, an EC50 value on a contrived HEK293 NRP2 over expression model has limited translational value to the in vivo biological reality of sarcoid patients. Worse, if the instrumentation and methods are the same between both experiments, it would suggest that the cell model has 30x the copies of NRP2 as the sarcoid patient estimates.
We would also add that this density of NRP2 benefits the fluorescence readout by virtue of avidity effects over affinity effects. Having two arms on efzo working together to bind a target and light up a sensor is different from reality where the avidity effect can’t come into play.
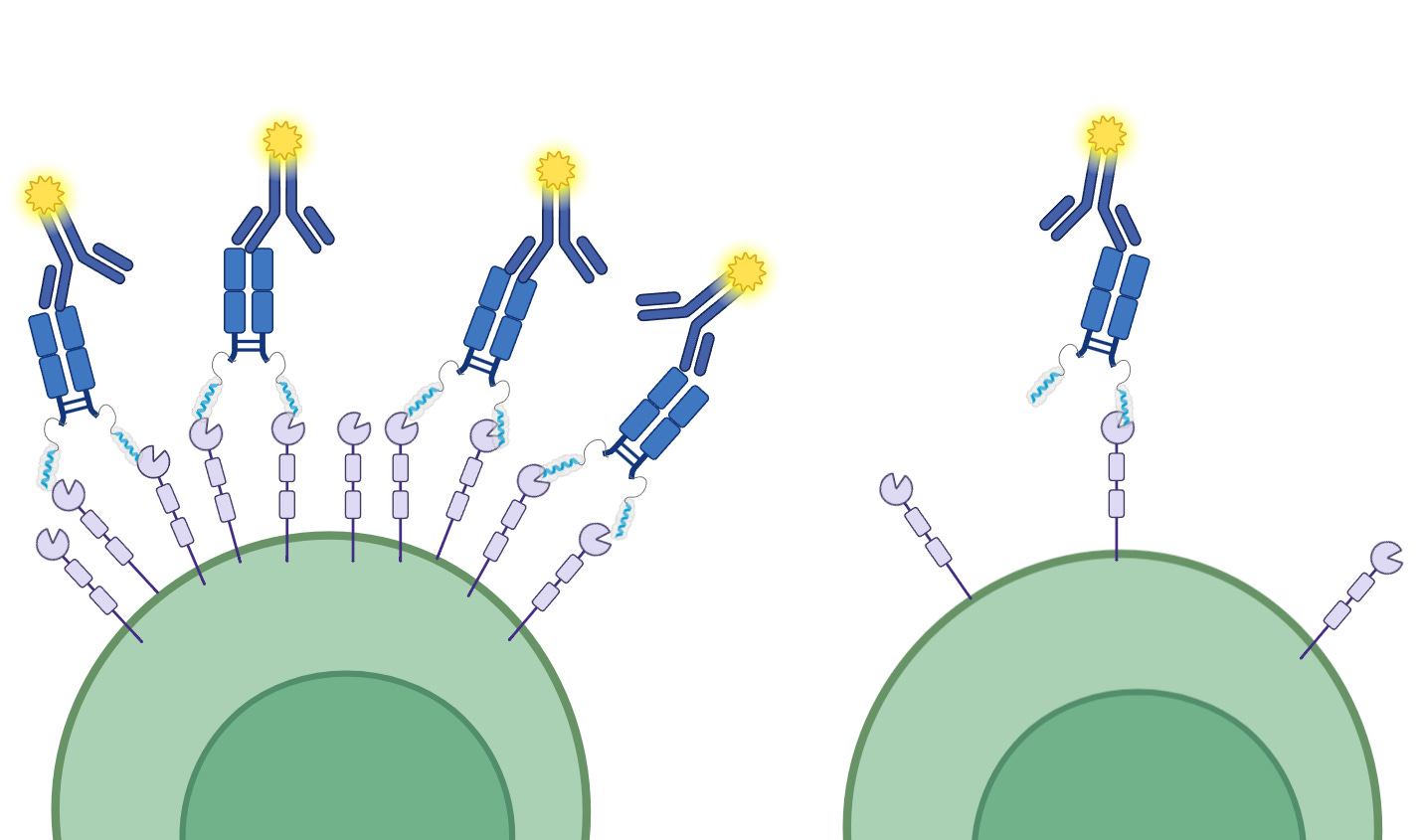
While it’s possible some avidity effects come into play in vivo due to NRP2 clustering on cell surfaces, we could not find evidence of this clustering phenomenon for alveolar macrophages, the sort aTyr claims to modulate with efzo. We see that it has been shown in other cell types but we also observe that not every copy of NRP2 is in such a cluster and much of the NRP2 within a granuloma would be inaccessible to efzo due to their density and cellular morphology, in the same way the core of solid tumor is inaccessible to therapeutics.
Affinity and target engagement
We note that the Science Translational paper VEGF-C positive control SPR experiment looks well done, as we have the multiple titrations of VEGF-C against NRP2.This shows the full association and dissociation curves for VEGF-C, and allows us to determine the KD value. A titration is required to generate an accurate KD model, because the mathematics require comparison to the RUmax against another concentration (see below) to build a curve. Also keep in mind, KD is a ratio of Kon and Koff, and different compounds can have very different Kon and Koff while having the exact same KD.

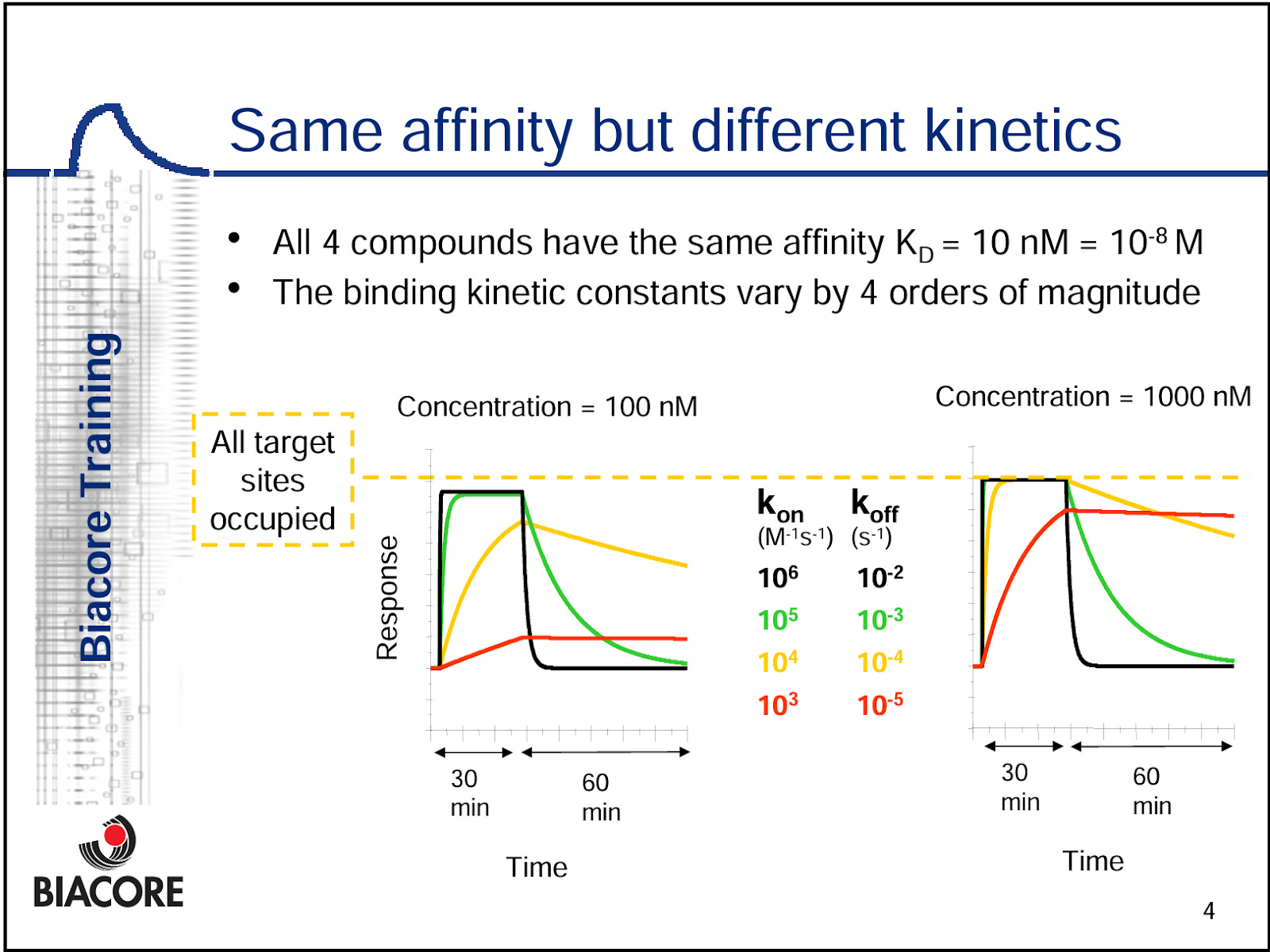

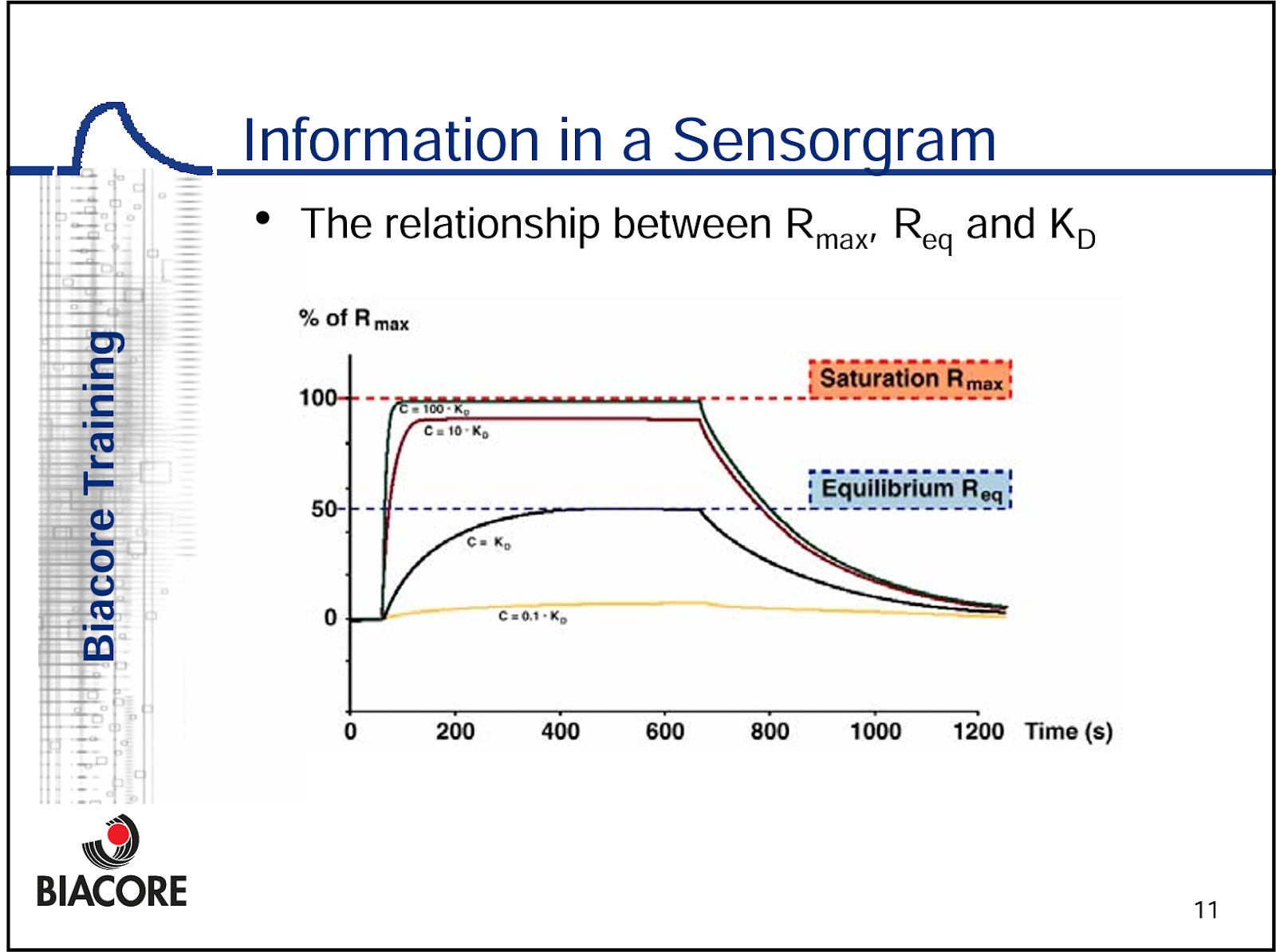
But for efzo, we have one sensogram shown, which means we can’t derive the actual KD value, and they don’t report KD anywhere in the literature or anywhere in their patents. They also made the choice to immobilize efzo to the sensor, and flowed over an Fc-tagged version of NRP2a2b1b2 domains. This is problematic because it is difficult to get 1:1 binding interactions with an immobilized bivalent molecule ligand, and this can be difficult to disentangle in the math. We would argue that, one ought to immobilize the NRP2 domain molecule(s) with a low enough concentration to mitigate avidity driven binding events, and flow over the fusion protein. Or, just flow over the HARSWHEP domain in this case. This ensures true 1:1 binding between the arms and an accurate KD value.


We find this experimental design and lack of reported KD bizarre for a lead molecule and indicative of a very low affinity. This puts into question the idea of meaningful target engagement, and we suspect there is very limited activity between efzo and NRP2 in vivo.
Epitope
We also note that the team never claimed an epitope at the amino acid level, but infer it from the binding domain surrogacy of their SPR experiments. They claim the HARSWHEP domain binds to only the NRP2 heterotrimer domain complex of a2b1b2, though b1b2 or b1 domains alone completely abrogate the binding. Perhaps this is a function of the epitope, it requires sufficient purchase on the heterotrimer to bind NRP2 though this is not reassuring given our concerns about binding affinity. Despite this, we did find an orientation in our simulations that satisfies this condition though as we said the binding affinity is dubious.
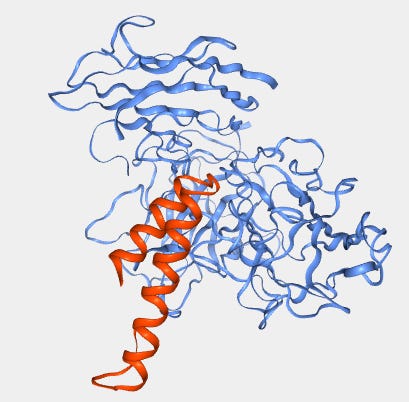
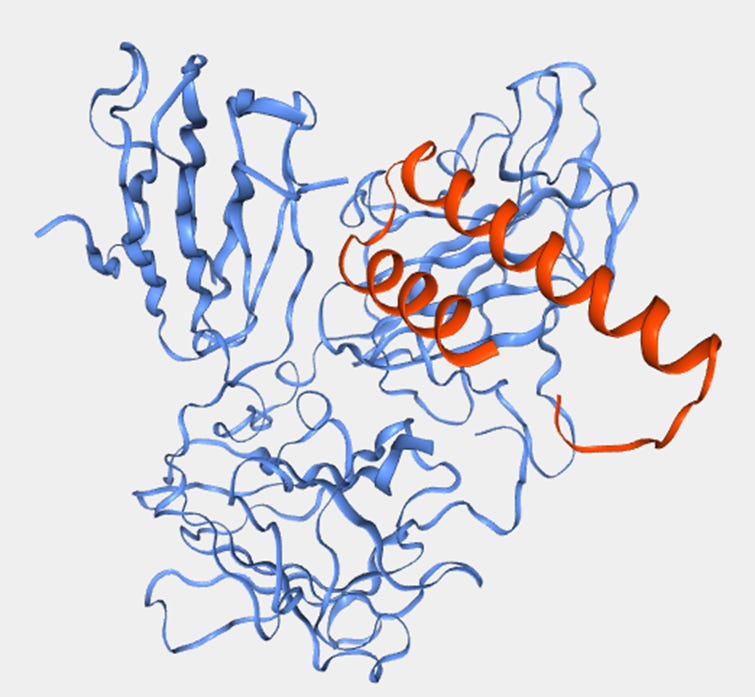
We also note that they could have performed other epitope mapping experiments such as cryoEM, hydrogen-deuterium exchange mass spectrometry, co-crystallization or site directed mutagenesis/ alanine scanning. This would of course offer the additional benefit of protecting the real estate on NRP2 for the aTyr team with a patent, which would seem valuable given that this binding site apparently does not interfere with VEGF or SEMA3F binding on NRP2. We wouldn’t expect many more functionally relevant epitopes to exist on NRP2 and protecting this one would seem prudent to avoid competition.
Finding KD and claiming an epitope is the norm in the biopharma industry, and even for the aTyr team as they did this workup for their anti-NRP2 antibodies21,22.
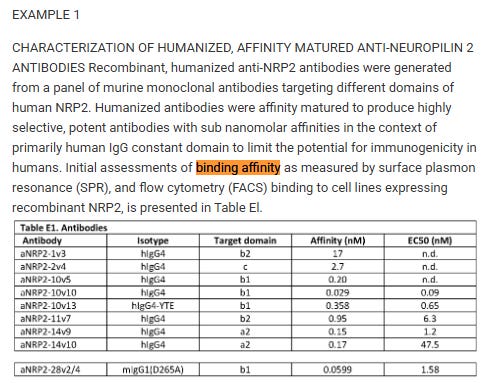
Again, we are troubled by the lack of epitope claims and lack of conclusive evidence proving the site or manner in which efzofitimod or the HARSWHEP domain interacts with NRP2.
Functional Proof for Mechanism of Action
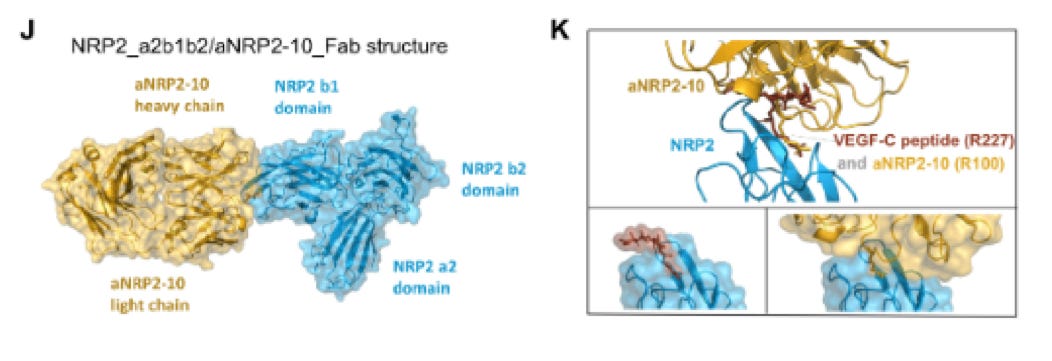
Delving more into the proposed mechanism of action- NRP2 agonism- we remain unconvinced that efzofitimod is agonizing the NRP2 receptor alone, or in tandem with its co-receptor(s) FLT4 and PLXNA1.
In the Science Translational paper, aTyr have shown a proximity dimerization assay which lights up when NRP2 and their co-receptors get close. We can see below that SEMA3F, VEGF-C both do this at their sub-saturation concentrations, which we assume are consistent with the reference provided to their antibody pre-clin workup (2 or 20nM for VEGF-C, 200nM for SEMA3F)18,21. Then, when they flow the efzo construct into the well which shows dimerization of the complex, it remains even when VEGF and SEMA3F are added. Thus the authors suggest that this proves they bind distinct epitopes compared to VEGF-C and SEMA3F.
This could be the case, it could also be the case that a low affinity binding molecule is stimulating some dimerization but is also kicked off once a high affinity binding molecule supplants it although the concentration differences could mitigate this. But we also find the concentrations used troubling- since VEGF-C was at either 2 or 20nM, it seems like there is much more efzo being used to potentiate FLT4 in almost all titrations. We also note the avidity effect can also be confounding this analysis, showing better performance in this cell model than it would otherwise show in sarcoid patient cells, and that endogenous VEGF-C and SEMA3F do not benefit similarly from this increased NRP2 expression due to 1:1 binding interactions.

Further, we are left wanting. We want to see a phosphorylation assay or other downstream assay proving that this potentiation of FLT4 and PLXNA1 lead to higher activity in some pathway once efzo is sitting on NRP2. Or that this lowers the bar for activation by the natural ligands. We have not seen any proof that dimerization leads to more downstream signalling and thus the MoA evidence is incompletely describing a physiological benefit to patients.
Does this alone prove efzo does nothing? Not exactly, as we have recently seen that obefazimod from Abivax has an incompletely described MoA and it still worked. However, we would point out that they had clean clinical data and demonstrable efficacy in pre-clin, and earlier clinical trials. If one is comfortable betting on a clinically confounded ph1/2, with a weak binding interaction, poorly understood target engagement and no functional pre-clinical proof for the MoA, then one has a risk tolerance much higher than the author.
Explaining NRP2 biology
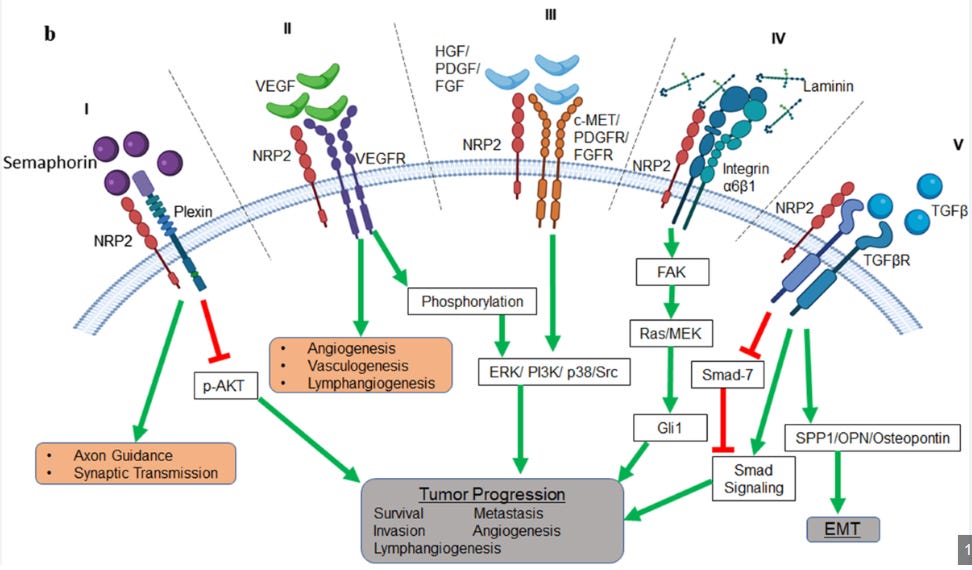
We’d like to spend a bit of time going over the biology of NRP2 in the human body.
From our reading, NRP2 is a double edged sword. In some cases, like acute injury, it can be anti-inflammatory and helpful in combating pro-inflammatory insults thru SEMA3F mediated signalling and by occupying NRP2 so VEGF can’t activate VEGFRs23,24. In other cases, particularly chronic cases, we have found evidence that it can actually stimulate a pro-inflammatory environment for tissue manifesting in the migration of monocytes and macrophages to tissues and promoting lymphangiogenesis via VEGF-C-VEGFR3 signalling and integrin mediated adhesion24,25.
The literature supports both cases, but they have different contexts. We propose that sarcoidosis is a complicated, heterogeneous disease with many pathways in play.
We know that VEGFR3 is overexpressed in the lung tissue of sarcoid patients, and VEGF-A/C/D are found to be elevated in the literature, and in the Adams biomarker poster26,27. It has been established that NRP2/VEGFR3 mediate lymphatic vessels sprouting in response to VEGF-C28,29. VEGF has been established to be immunosuppressive in a variety of contexts, and we would argue that this extends to sarcoidosis patients as well 30.
In fact, research shows that VEGF-A can act on NRP1 to suppress DC maturation, preventing upregulation of MHCII and lowering the secretion of IL-1b, IL-6, IL-12 and TNF-a31. This lowering of DC maturation would also indirectly lower T-cell driven IFN-γ and IP-10 expression via lowered DC → Th1 stimulation. Further, VEGF-C/VEGFR-3 mediated lymphatic endothelial cell (LEC) recruitment can also drive this Th1 response on NRP2 by promoting immune tolerance through PD-L1–dependent deletion/anergy and antigen presentation without costimulation32,33.
Thus VEGF-A/C/D can drive an immunosuppressive phenotype, which could explain some of the observed cytokine results in the pre-clinical and clinical studies.

We see above that the company purports a clinical benefit to the reduction of immune activity. We would argue that this is not very relevant, given the multitude of failed MoAs suppressing immune function and in this case the return of sarcoid disease biomarkers -despite the different baselines noted previously- are much more relevant.
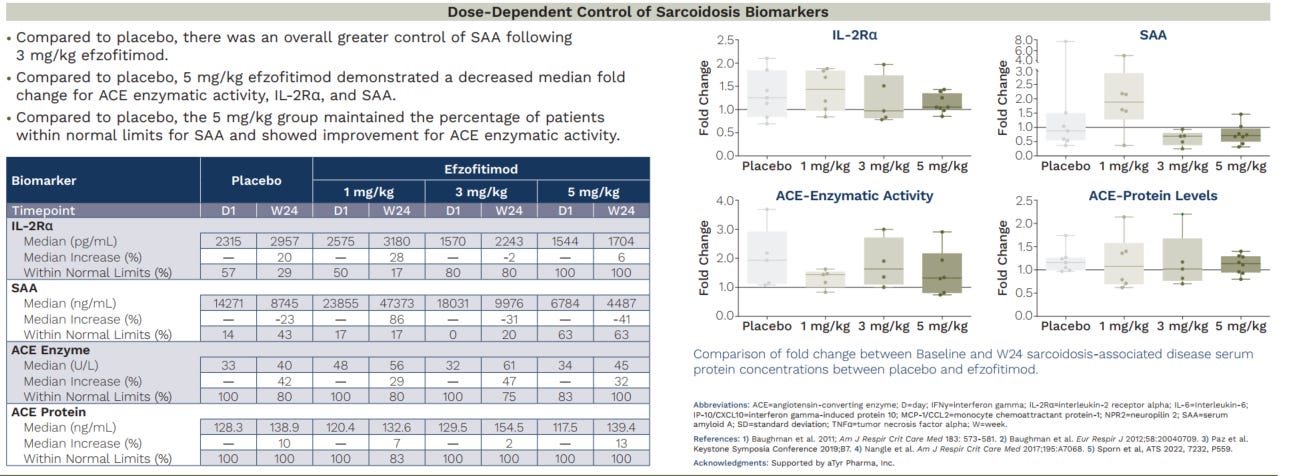
We note that the median ACE enzyme activity and serum protein levels in particular are rising in all arms (and add that we would love to see the mean). This is important to observe since ACE is a surrogate for granuloma activity. We don’t need to worry about ACEi drugs interfering with this signal, as they are removed from the analysis. Similarly IL2Ra is rising in all arms, again showing return of disease activity.
To be clear- here we are saying that as a marker of disease activity (specifically granuloma activity)- not an aggregate state- we see disease progression on these markers. This data suggests that efzofitimod confers no protective benefit against granuloma activity compared to placebo even when starting from a healthier disease state.
To go further, we posit that this is a sign that the VEGF-C/D/VEGFR-3 mediated lymphangiogenesis could stimulate disease progression though we lack experimental evidence for this claim.
Upon examination of the AE table in the Chest paper we see many manifestations of disease progression (cough, fatigue, dyspnea) listed as AEs in the patients taking the drug. We acknowledge that this could be a noisy signal resulting from steroid withdrawal but given the baseline imbalance and higher number of patients in the placebo arm without so many disease manifestations we are keeping a close eye on the progress of patients in ph3.

The missing link in this theory would be: where is VEGF signalling coming from? And as we established previously, we put forth that efzofitimod’s binding domains (HARSWHEP2-60) could have some affinity for CCR5 bearing lymphocytes, drawing them to lung tissue and amplifying the local VEGF mediated signalling and immunosuppression while simultaneously promoting lymphangiogenesis in endothelial tissue. This is supported by computational simulations between HARS WHEP and CCR5. This is supported by the Howard et al paper. These arguments are not refuted by a lack of subsequent study.
Final review of attempts in sarcoidosis
Lastly, further investigation into the literature has shown that similar alveolar macrophage driven mechanisms of action that efzo is trying to work through have failed to benefit patients34. This includes the anti-IL1 class, and TNFa class which have had outright failures or mixed results. We note that there failures in downregulating the pathways that aTyr highlights are indicative of efficacy, such as IL-6.
On the whole, this suggests that cytokine suppression alone is insufficient to reduce disease burden. Indeed, as many have noted, sarcoidosis is a very heterogeneous disease likely with multiple etiologies none of which seem to be completely understood. To suggest that NRP2 is the master pathway that will benefit all patients uniformly seems to us unlikely.
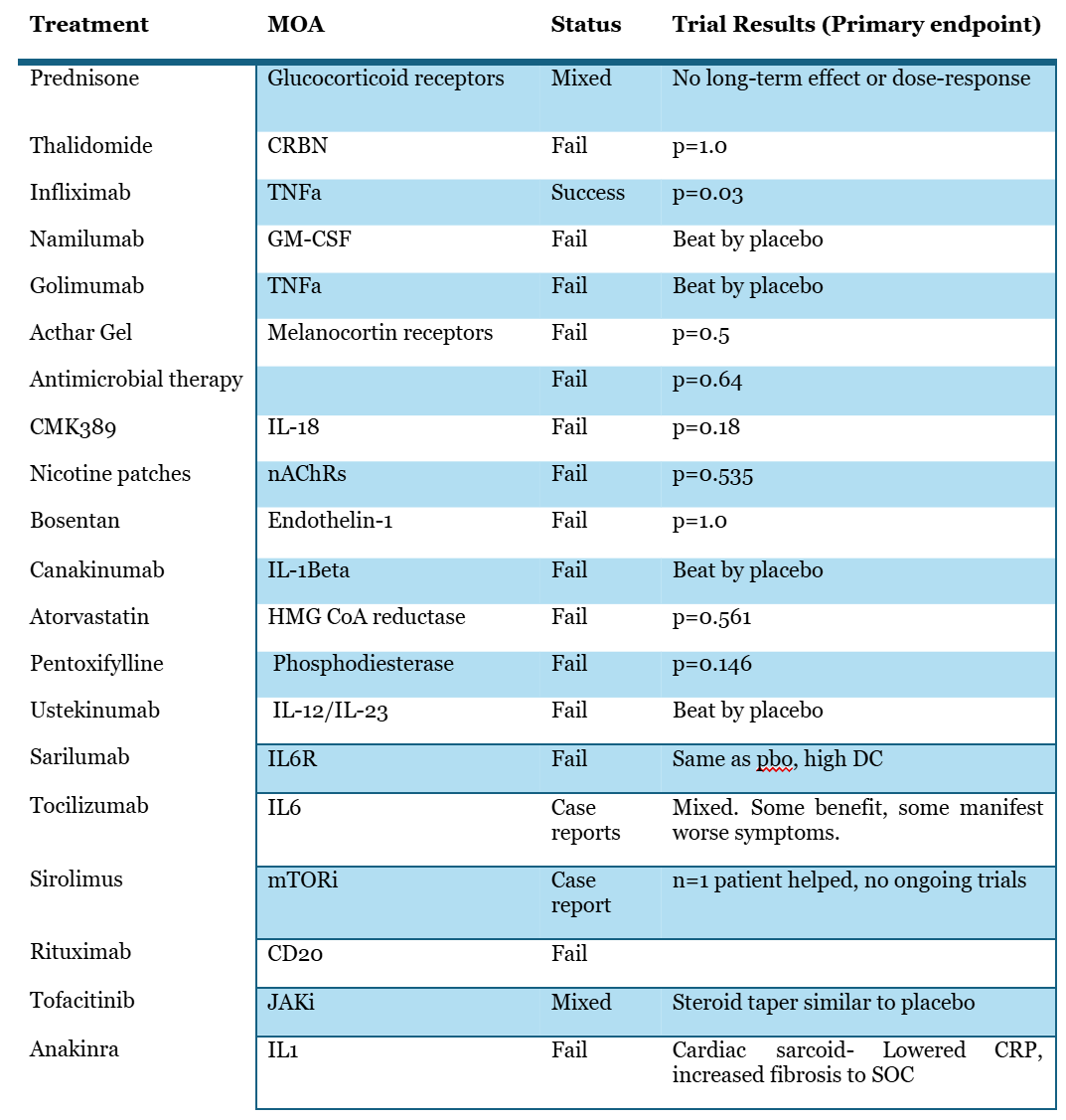
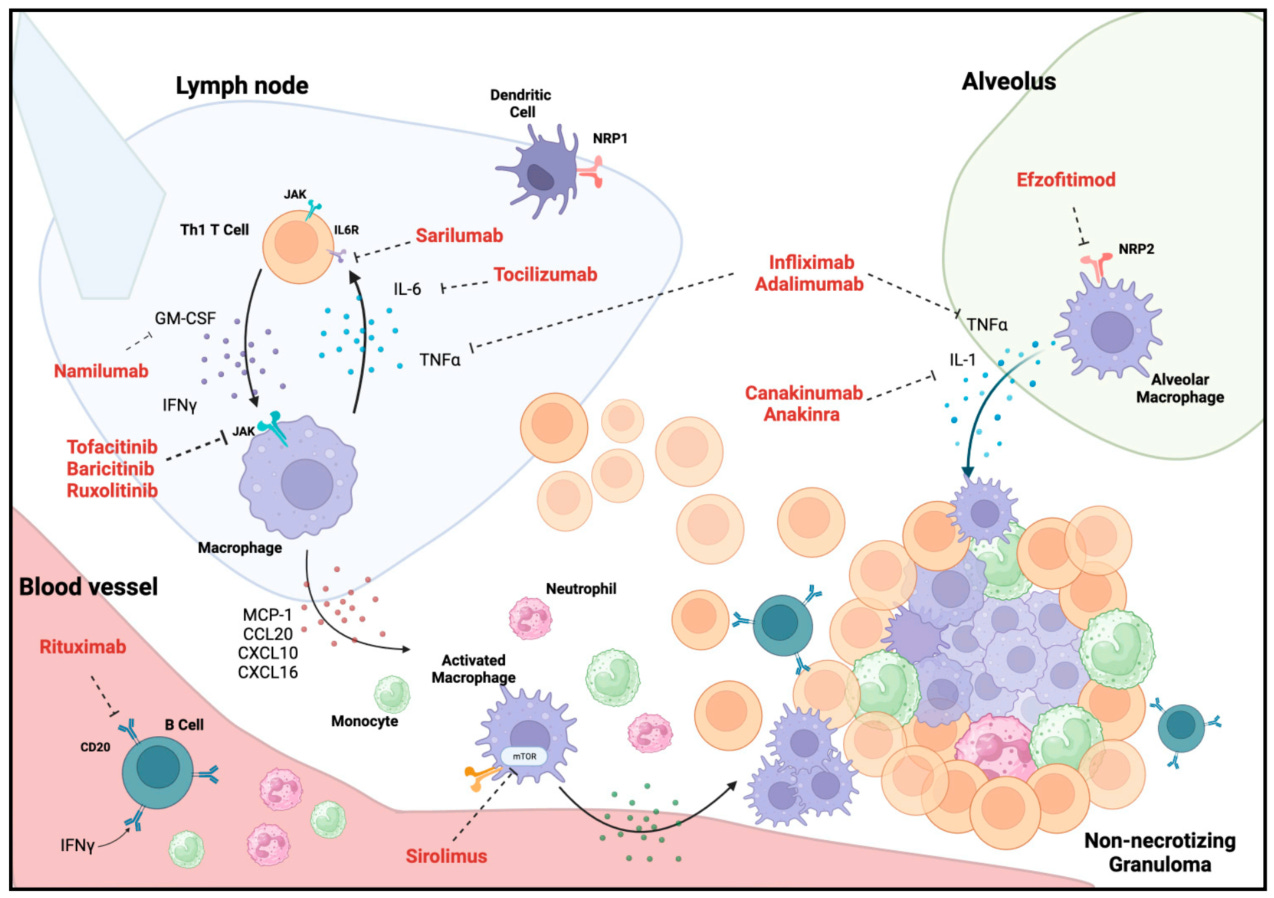
Pre-clinical data
Some have said the pre-clinical data examined in our first report was “selective”. We disagree, and counter that the data presented by aTyr to establish activity in its pre-clinical models of sarcoid is selective. We reject the notion that bleomycin murine models are superior to the P. Acnes models in simulating sarcoid, on the grounds that they fail to recapitulate the basic cellular morphology of the disease seen in sarcoid patients, and, because P. acnes has been proposed as a causal agent in the formation of sarcoidosis granulomas in patients. Bleomycin may be a more appropriate model for fibrotic tissue injury like IPF or even much later stage sarcoid, but for granulomatous tissue formation this is an inappropriate surrogate for disease.
Further, experts do not agree that it is an appropriate pre-clinical model to mimic the state of sarcoid patients as we see Besnard and Jeny discuss in their paper “Are there Good Models of Sarcoidosis?”35. While there is no perfect model they do acknowledge that the P. Acnes model does match some of the state observed in patients with sarcoidosis such as granulomatous tissue formation, a similar cytokine profile, and a reasonable etiology of disease commensurate with patient exposure. They do not mention bleomycin models in the paper.
The P. acnes induced murine models used for efzo show that it is an ineffective agent in the treatment of sarcoid. We see that in their Science Translational paper:

Ashcroft score is barely reduced (within the error bars), and granulomatous inflammation is similarly, barely reduced.
We see in their 2019 poster where a lower dose seems to increase Ashcroft score over vehicle and a higher dose has a little effect. A reduction in inflammatory cytokines is irrelevant when the disease is progressing.

Our highlighting of the only functional pre-clinical model is not “being selective”, it is being objective in assessing the low activity of efzofitimod in treating the causal mechanisms driving disease in sarcoidosis. Inflammatory biomarkers don’t matter if granulomas persist and thrive in vivo, which we also see in the clinical biomarkers from patients.
Concluding remarks
After a review of the literature we find that patients vary in disease manifestation and progression. It is difficult to pin down the disease state and assess uniform outcomes for patients. That said, it is helpful to have a similar study protocol to look back on and guide us towards the question the current ph3 study is trying to answer: can patients taper off steroids quicker than the guidelines suggest?
To that, we would say yes. When it comes to the clinical trial and how efzo will compare to placebo, we think they will perform quite similarly, with roughly 70-75% of patients in all arms being off steroids between weeks 45-48 of the study.
We believe this, because as we detail further, we do not find the rationale for the MoA of efzofitimod compelling. We lack fundamental understanding of target engagement via KD, and deduce that the EC50 values given by the company overstate the activity of the drug due to avidity effects on cell models overexpressing NRP2 beyond physiologically relevant conditions. The lack of discrete amino acid level binding epitope and interaction mechanisms to NRP2 are troubling. We have not seen any downstream proof of agonism via NRP2 through FLT4 or plexin or any of the co-receptors of NRP2, thus without evidence for this claim we are unconvinced it is modulating NRP2 to any effect.
In reviewing NRP2 biology further we see that it is a double-edged sword which can reduce or promote an inflammatory condition locally. The finding of VEGF-C/D/VEGFR-3 driven lymphangiogenesis potentially leading to further granuloma development and an immunosuppressive phenotype does not engender confidence in this MoA. The possibility that the HARSWHEP2-60 domain is recruiting CCR5 remains to be validated experimentally, but our computational simulations suggest the Howard paper could be replicated. This would also help to explain the observed reduction in cytokines.
Our re-review of the pre-clinical models suggest that not only was our initial observation of the poor outcomes of the sarcoidosis specific models correct, it is in-line with expert researchers in the field for assessing the translational value of these model systems. Thus, we have very little reason to believe that efzofitimod can translate poor pre-clinical work into successful clinical results.
We attribute the differences in placebo adjusted outcomes from the ph1/2 to baseline imbalances between groups. It seems that these may stem from the MAD clinical trial design, and the post-hoc observation that more patients tapered to 0mg/d in the 5mg/kg arm is spurious due to the 2:1 dosing regimen and lack of equipoise across all dose cohorts.
References
2. Culver, D. A. et al. Efzofitimod for the Treatment of Pulmonary Sarcoidosis. Chest 163, 881–890 (2023).
3. Kahlmann, V. et al. First-Line Treatment of Pulmonary Sarcoidosis with Prednisone or Methotrexate. N. Engl. J. Med. 393, 231–242 (2025).
4. Gottlieb, J. E., Israel, H. L., Steiner, R. M., Triolo, J. & Patrick, H. Outcome in Sarcoidosis. Chest 111, 623–631 (1997).
5. Baughman, R. P. Presenting characteristics as predictors of duration of treatment in sarcoidosis. QJM 99, 307–315 (2006).
6. Bilgin, B., Bilgin, M. K., Erol, S., Celik, G. & Ozdemir Kumbasar, O. Prognosis of sarcoidosis and factors affecting prognosis. Sarcoidosis Vasc. Diffuse Lung Dis. 40, e2023054 (2023).
7. Partin, M., Clebak, K. T., Chen, R. & Helm, M. Sarcoidosis: Evaluation and Treatment.
8. Stadnyk, A. N., Rubenstein, I. & Rosenthal, T. Clinical features of sarcoidosis in elderly patients. Sarcoidosis 5, 121–3 (1988).
9. Johns, C. J. A TEN YEAR STUDY OF CORTICOSTEROID TREATMENT OF PULMONARY SARCOIDOSIS.
10. Baughman, R. P. et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur. Respir. J. 58, 2004079 (2021).
11. Broos, C. E. et al. Daily home spirometry to detect early steroid treatment effects in newly treated pulmonary sarcoidosis. Eur. Respir. J. 51, 1702089 (2018).
12. Dhooria, S. et al. High-dose (40 mg) versus low-dose (20 mg) prednisolone for treating sarcoidosis: a randomised trial (SARCORT trial). Eur. Respir. J. 62, 2300198 (2023).
13. Fang, C., Zhang, Q., Wang, N., Jing, X. & Xu, Z. Effectiveness and tolerability of methotrexate in pulmonary sarcoidosis: A single center real-world study. Sarcoidosis Vasc. Diffuse Lung Dis. 36, 217–227 (2019).
14. Gavrysyuk, V. et al. Efficacy and Tolerability of Methotrexate and Methylprednisolone in a Comparative Assessment of the Primary and Long-Term Outcomes in Patients with Pulmonary Sarcoidosis. Diagnostics 11, 1289 (2021).
15. Walker, G. et al. Exposure-response analyses of efzofitimod in patients with pulmonary sarcoidosis. Front. Pharmacol. 14, 1258236 (2023).
16. Howard, O. M. Z. et al. Histidyl–tRNA Synthetase and Asparaginyl–tRNA Synthetase, Autoantigens in Myositis, Activate Chemokine Receptors on T Lymphocytes and Immature Dendritic Cells. J. Exp. Med. 196, 781–791 (2002).
17. Norden, D. M., Navia, C. T., Sullivan, J. T. & Doranz, B. J. The emergence of cell-based protein arrays to test for polyspecific off-target binding of antibody therapeutics. mAbs 16, 2393785 (2024).
18. Nangle, L. A. et al. A human histidyl-tRNA synthetase splice variant therapeutic targets NRP2 to resolve lung inflammation and fibrosis. Sci. Transl. Med. 17, eadp4754 (2025).
19. Paz, S. et al. Neuropilin-2, the Specific Binding Partner to ATYR1923, Is Expressed in Sarcoid Granulomas and Key Immune Cells. in B35. SARCOIDOSIS CLINICAL AND MECHANISTIC STUDIES A3099–A3099 (American Thoracic Society, 2020). doi:10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A3099.
20. Baughman, R., Walker, G. & Culver, D. Efzofitimod: A Novel Anti-Inflammatory Agent For Sarcoidosis. SARCOIDOSIS Vasc. DIFFUSE LUNG Dis. 40, (2023).
21. Xu, Z. et al. Inhibition of VEGF binding to neuropilin-2 enhances chemosensitivity and inhibits metastasis in triple-negative breast cancer. Sci. Transl. Med. 15, eadf1128 (2023).
23. Nakayama, H. et al. Regulation of mTOR Signaling by Semaphorin 3F-Neuropilin 2 Interactions In Vitro and In Vivo. Sci. Rep. 5, 11789 (2015).
24. Dhupar, R. et al. Orchestrating Resilience: How Neuropilin-2 and Macrophages Contribute to Cardiothoracic Disease. J. Clin. Med. 13, 1446 (2024).
26. Yamashita, M. et al. Heterogeneous Characteristics of Lymphatic Microvasculatures Associated with Pulmonary Sarcoid Granulomas. Ann. Am. Thorac. Soc. 10, 90–97 (2013).
27. Adams, R. et al. Efzofitimod (ATYR1923) Treatment Reduces Pro-inflammatory Serum Biomarkers in Pulmonary Sarcoidosis Patients.
28. Xu, Y. et al. Neuropilin-2 mediates VEGF-C–induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 188, 115–130 (2010).
29. Islam, R. et al. Role of Neuropilin-2-mediated signaling axis in cancer progression and therapy resistance. Cancer Metastasis Rev. 41, 771–787 (2022).
30. Ribatti, D. Immunosuppressive effects of vascular endothelial growth factor (Review). Oncol. Lett. 24, 369 (2022).
31. Oussa, N. A. E. et al. VEGF Requires the Receptor NRP-1 To Inhibit Lipopolysaccharide-Dependent Dendritic Cell Maturation. J. Immunol. 197, 3927–3935 (2016).
32. Garnier, L., Gkountidi, A.-O. & Hugues, S. Tumor-Associated Lymphatic Vessel Features and Immunomodulatory Functions. Front. Immunol. 10, 720 (2019).
33. Lucas, E. D. & Tamburini, B. A. J. Lymph Node Lymphatic Endothelial Cell Expansion and Contraction and the Programming of the Immune Response. Front. Immunol. 10, 36 (2019).
34. Nelson, N. C., Kogan, R., Condos, R. & Hena, K. M. Emerging Therapeutic Options for Refractory Pulmonary Sarcoidosis: The Evidence and Proposed Mechanisms of Action. J. Clin. Med. 13, 15 (2023).
35. Besnard, V. & Jeny, F. Models Contribution to the Understanding of Sarcoidosis Pathogenesis: “Are There Good Models of Sarcoidosis?” J. Clin. Med. 9, 2445 (2020).
Excellent work, I really appreciate what you do!