

Company and Platform
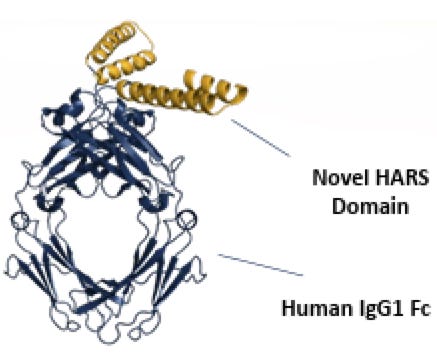
aTyr Pharma (NASDAQ:ATYR) is a clinical‐stage biotherapeutics company founded in 2005 by Drs. Paul Schimmel and Xiang‑Lei Yang, pioneers in the field of tRNA synthetases. Leveraging a global patent estate around these enzymes, ATYR’s flagship candidate, efzofitimod (ATYR1923), is a splice‑variant fragment of histidyl‑tRNA synthetase (HARS) purported to agonize neuropilin‑2 (NRP2) and modulate immune responses in interstitial lung diseases, including pulmonary sarcoidosis.
Scientific Rationale and Mechanism of Action
Efzofitimod was shown to bind NRP2 via a Retrogenix screen rather than through a target‐driven approach. ATYR hypothesizes that NRP2 agonism downregulates pro‑inflammatory cytokines, thereby reducing granuloma formation and fibrosis. Contradictorily, HARS fragments have been found to act as chemotactic cytokines for CCR5‐bearing cells, a pro‑inflammatory pathway that may counteract the intended anti‑inflammatory effect. And many anti-inflammatory agents have been tried in sarcoid yielding little long term benefit for patients including the first-line therapy, prednisone.
Preclinical Evidence
In vitro affinity studies demonstrate efzofitimod binds NRP2 with a sub‑30 nM EC₅₀ and exhibits reasonable pharmacokinetics (t½: 9–11d). In murine models of lung injury and granulomatous inflammation, efzofitimod reduced inflammatory biomarkers but failed to demonstrate clear reductions in granuloma burden, raising questions about translatability to human disease.
Phase 1/2 Clinical Data (NCT03824392)
A randomized, placebo‑controlled study enrolled 37 sarcoidosis patients across three dose cohorts (1, 3, 5 mg/kg). Primary endpoints focused on safety, corticosteroid tapering, and functional lung assessments at week 24 :
Baseline Imbalances: Higher‑dose cohorts had more favorable FVC% and biomarker profiles at baseline, confounding the interpretation of results.
Steroid Tapering: No dose arm exhibited improvements in maintaining prednisone reductions versus placebo; the observed 1.8 mg/day placebo‑adjusted reduction in the 5 mg/kg arm falls well within standard deviation.
Lung Function (FVC): Modest, non–dose‑dependent changes were observed; comparisons with infliximab trials highlight efzofitimod’s limited impact on objective functional endpoints.
Post‑hoc Bayesian and ANCOVA analyses accounting for baseline differences suggest that placebo‐adjusted effects are likely driven by initial cohort imbalances rather than a true effect by the drug on the disease.Phase 3 Outlook (NCT05415137)
The upcoming 268‑patient trial will assess 3 mg/kg and 5 mg/kg dosing with a revised primary endpoint: mean change in daily oral corticosteroid dose during Weeks 45–48. Statistical significance will require rigorous control of alpha across two dose arms (p < 0.025 or a Holm split). Given the corrected baseline imbalances and milder patient population, we believe replicating the modest Phase 1/2 placebo adjusted steroid reductions will be a tough task. Even if efzo does, we question the clinical significance of a 2-3mg/d reduction in OCS in a patient population where spontaneous disease resolution is very common.
Risks and Considerations
Novel Target Uncertainty: Limited understanding of NRP2 biology and contradictory CCR5 chemotactic data warrant considerable caution.
Clinical Signal Strength: Preclinical granuloma data and Phase 1/2 outcomes show very weak translational efficacy.
Statistical Hurdles: Baseline normalization and stringent multiplicity controls significantly raise the bar for Phase 3 success.
Trial Design: Primary endpoint may be unreachable and or not clinically meaningful, taking into account prednisone’s lack of long-term efficacy in sarcoidosis as noted in a Cochrane review
Financial Position
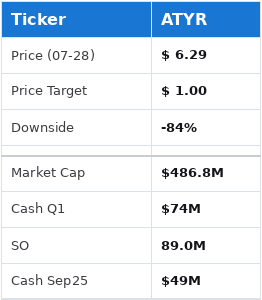
Given that the company holds $74 million in cash with 89 million shares outstanding, and a burn rate of $15 million/quarter, we see the cash value of the company at roughly $0.55 going into September. With a failure in this upcoming ph3 we believe it would be both hard and illogical for the stock to trade well above this valuation.
Conclusion
While ATYR’s platform represents a new application of tRNA synthetase biology, the unconventional discovery path, the fate previously met by related mechanisms in clinical trials, conflicting mechanistic evidence, modest early clinical signals, as well as a questionable clinical trial design cast significant doubt on efzofitimod’s ability to meet Phase 3 endpoints or offer benefit to patients.

Disclosures: This Report is provided by Fourier Transform Research solely for educational purposes and constitutes impersonal, generic commentary based entirely on publicly available data. The views expressed herein reflect the personal opinions of the author as of the date of publication; they are not tailored to any individual’s financial situation or objectives and do not constitute investment advice, a recommendation, or an offer to buy or sell any security. The author holds a short position in the securities discussed. No part of the author’s compensation is, was, or will be directly or indirectly related to any specific recommendation or view contained herein. The author has not received—and will not receive—any payment of any kind from the company(ies) whose securities are covered by this Report. This Report contains no material non‑public information. Trading the securities covered herein is subject to a blackout period for the author commencing 72 hours before and ending 24 hours after the Report’s public release. All factual data are believed to be accurate at the time of writing, but Fourier Transform Research makes no warranty as to its completeness or accuracy; recipients should conduct their own due diligence. Past performance is not indicative of future results.
Introduction to company and tRNA Synthetases
aTyr Pharma was founded in 2005 in San Diego, CA by Paul Schimmel and Xiang-Lei Yang who were conducting research on tRNA synthetases at Scripps. Paul remains on the board to this day, and so does the work he pioneered in this field. They have been trying to turn these tRNA synthetases into useful drugs for a long time now and, from our point of view, that’s a bit of an odd way to go about drug discovery.
What do we mean by that?
Well, the author has 8+ years of antibody drug discovery experience and typically the process looks something like: I have an interesting target and want to make a drug against it. Whether I want to inhibit it, agonize it, recruit some other cells or signal using that target is up for later development but usually you start with a target and find a way to drug it as Suzuki et al. 2015 showed us by describing the mechanisms of action of therapeutic antibodies in their review1.

When looking into efzofitimod, we were surprised to find out that the company had developed this molecule and only then found out it hit NRP2 on a cross-reactivity test, the Retrogenix assay. Usually you take a lead molecule and run it on this assay to see if it hits against anything other than your target of interest. You do this because you want to know whether you’ll have some off-target effect you didn’t plan on seeing that could have an unintended side effect in the clinic. But this was actually how aTyr decided to run with efzofitimod into sarcoidosis in the first place.
NRP2 is a novel target. There are very few studies on it compared to other hot areas in biology. This creates some of the typical “novel target” problems like understanding how it works, what it does, and how it relates to disease. Through our research we’ve been able to answer some of those questions as we’ll show, but for now we’ll establish firmly that this is not a straightforward target and the literature does not inform sufficiently about it.
With that in mind we will also point out that the company has a history of failure in the clinic. Assets like ATYR1940, the ORCA program, and ATYR2810 have all been killed. ATYR1940 (Resolaris) interestingly was a kind of version of efzo, another HARS (human histidyl tRNA synthetase) molecule that they tried to use on adolescent boys with Facioscapulohumeral Muscular Dystrophy (FSHD).
As we can read from the Schimmel group, the original theory for the WHEP domain MoA surrounded its use as a chemotactic agent for lymphocytes in muscle disease2.
“Possibly, HisRS (Histidyl tRNA synthetase) and its two WHEP domain–containing SVs are involved in maintaining immune homeostasis in muscle. When immune surveillance or clearance is needed, HisRS proteins attract immune cells to muscle tissue. In support of this hypothesis, the N‑terminal WHEP domain of HisRS may be chemotactic for lymphocytes and activated monocytes. Possibly because of their persistent presence, in DM patients, the HisRS proteins are eventually seen as “foreigners,” and autoantibodies against HisRS, especially the WHEP domain, are generated. These autoantibodies may antagonize the immune homeostatic role of HisRS proteins and gradually lead to myositis.”
Further, the idea with these tRNA synthetase fragments was rooted in the observation that certain patients with inflammatory myopathies such as dermatomyositis and polymyositis develop autoantibodies against histidyl‑tRNA synthetase (anti‑Jo‑1). This suggested that the HisRS protein, particularly its WHEP domain, plays an active role in muscle inflammation, likely acting as a chemokine to recruit immune cells. This led the team to target patients with diseases related to muscle inflammation like FSHD and LGMD. Interestingly, rather than trying to block this chemotactic signal that is over-active in these patients, the company tried supplementing it with pretty much full length HisRS. Patients developed anti-drug antibodies, not unlike anti-Jo-1, and the program was terminated for lack of efficacy.
Beyond this, they also tried to run a discovery program for cancer (ORCA), but couldn’t find any candidates, killed the program and laid off 30% of the staff. ATYR2810 was an interesting molecule- an antibody against NRP2 but as an antagonist, so the opposite of what Efzo aims to be (an agonist). We find this interesting because they are kind of hitching their wagon to this NRP2 molecule after happening upon it on the Retrogenix assay some years ago.
It’s worth saying tRNA synthetases are a kind of weird set of molecules too. They are not really made to act on classical proteins that regulate disease. They are really meant to be, in the words of CEO Sanjay Shukla on BiotechTV, an enzyme that helps a tRNA conjugate to a particular amino acid and shepherd it to help it make a protein.

They are, strictly speaking, not meant to act upon proteins like NRP2 or anything else really, as far as we understand them. But Dr. Schimmel has put forth that fragments of these molecules are relevant to the immune system3. In the current case, specifically by agonizing (turning up) NRP2 and dampening inflammation in sarcoidosis4. This is of course a bit of a switch from the ATYR1940 story which was: let’s give full length HARS to patients and see if that helps with muscular dystrophy. That program was stopped due to immunogenicity and lack of efficacy as per their 2017 10K. Now they have pulled out a HARS fragment, coupled it to a human antibody Fc, and made efzo, after finding out that it happened to hit NRP2.
Coupling anything to an Fc is a good way to make almost anything more drug-like. It will improve PK (half-life), help reduce immunogenicity, and enhance distribution throughout the body. If you have something that binds to a target, slapping it on an Fc is a great way to get data to run into a ph1.
When we look at these facts and this timeline we’re really struck by the fact that this is just not how R&D normally (ever?) goes for a biologic drug candidate. We’re not talking about a small molecule where we test dirt on 1000 targets and see what turns up. To us, this isn’t really building a drug for a disease as much as finding a disease for a drug. But it’s happened before, like in Viagra (a small molecule), so we won’t say that this fact on its own disproves anything. We’re just saying that this is not how we normally go about drug discovery, particularly with proteins.
Lastly, we think the following excerpt from the August 2Q2018 10Q highlights some facts around this narrative:
Aug 2018 "We are a biotherapeutics company engaged in the discovery and development of innovative medicines based on novel immunological pathways. We have concentrated our research and development efforts on a newly discovered area of biology, the extracellular functionality of tRNA synthetases. Built on more than a decade of foundational science on this novel biology and its effect on immune responses, we have built a global intellectual property estate directed to a potential pipeline of protein compositions derived from 20 tRNA synthetase genes. We are focused on the therapeutic translation of the Resokine pathway, comprised of extracellular proteins derived from the histidyl tRNA synthetase (HARS) gene family, one of the tRNA synthetase genes. Our clinical-stage product candidate, ATYR1923, is based on the Resokine pathway, binds to the neuropilin-2 receptor and is designed to down-regulate immune engagement in interstitial lung diseases and other immune-mediated diseases. In parallel, we have also been expanding our knowledge base of the therapeutic potential of ATYR1923 by conducting several in vivo and in vitro models to further elucidate its potential clinical utility as an immuno-modulator. For example, we have presented the positive results of ATYR1923 in a mouse bleomycin lung injury model and a rat bleomycin lung injury model at the 2017 and 2018 American Thoracic Society Annual Meetings, respectively. In addition, we presented positive findings of ATYR1923 in a scleradermatous chronic graft versus host disease model at the Scleroderma Foundation’s 2018 National Patient Conference. These data, as well as the Phase 1 clinical trial results, will help inform selection of the indication for future clinical trials for our ATYR1923 program. At this time, we plan to initiate a multi-ascending dose, placebo-controlled Phase 1b/2a study in patients with interstitial lung disease in the fourth quarter of 2018.
What are they telling us here?
We have a platform built on tRNA synthetases
A patent estate around them and we found a place to plug that IP into.
Looks good in ILD models, we’re trying that indication first.
No mention of sarcoid at all.
This was when they decided to first push efzofitimod into the clinic. Platforms were very hot at the time, and, from that perspective this made sense. But we also think it’s interesting that they didn’t focus on or mention sarcoid explicitly at this time, again making us wonder if they were not simply searching for an indication for this drug. There’s something to be said about having multiple shots on goal, for a single asset, with respect to multiple hypothesis testing- eventually, you might get lucky.
Disease History
With that history lesson behind us, let’s look at the focus of the upcoming readout: pulmonary sarcoidosis.
A brief introduction from our AI friends:
Pulmonary sarcoidosis is a form of sarcoidosis, an inflammatory disease characterized by the formation of granulomas—small clumps of immune cells—in the lungs and sometimes other organs. It primarily affects the lungs, causing symptoms like persistent dry cough, shortness of breath, chest pain, or wheezing. Some patients may experience fatigue, fever, weight loss, or night sweats. The exact cause is unknown, but it’s thought to involve an abnormal immune response, possibly triggered by environmental factors, infections, or genetics.
The disease varies widely: some individuals are asymptomatic, while others develop severe complications like pulmonary fibrosis, where lung tissue scars and stiffens, impairing breathing. Diagnosis typically involves chest X-rays, CT scans, lung function tests, or biopsies to confirm granulomas. Treatment may not be needed for mild cases, but corticosteroids, immunosuppressive drugs, or biologics like anti-TNF therapies are used for severe or progressive disease to manage inflammation and prevent complications.
About 20-50% of cases resolve spontaneously within a few years, but chronic or progressive disease can lead to long-term lung damage. It’s more common in adults aged 20-40, with higher prevalence in African Americans and Northern Europeans. Always consult a healthcare provider for personalized information and management.
Few key points here:
Formation of granulomas in the lungs
Exact cause unknown
Treatment not needed for mild cases
Treatment needed for severe or progressive disease
20-50% of cases resolve spontaneously in a few years
Verify these claims for yourself with the guidelines established by the ATS5 and highlighted by Soto-Gomez, 2016:

We can see from the above table that clearly, staging matters a lot in this disease, as rates of spontaneous resolution can be incredibly high in mild disease ranging from 40% to 90%(!) for radiologically tested stage I/II patients. This is a critical point to appreciate, because mild patients can very often have disease resolution without drug. This was also noted by Culver in the most recent UpToDate publication on Sarcoid early (<5yr) treatment6:


In concluding remarks they are saying that 75% of patients do not need treatment and have spontaneous disease resolution. Is there really a place for efzo in this paradigm?
What about treatments?

Front line therapy is corticosteroids, and we also see some other agents being used in later lines like methotrexate and the biologics like infliximab5. However, we should note that in a Cochrane meta-analysis over 13 studies on over 1000pts treated with OCS for 6 to 24mths authors found no long term benefits to mortality, lung function, radiologic findings or disease progression, sadly7.
There may be a benefit to patients initially, but conflicting evidence would suggest that this effect may be transient and may not hold up long term. Near term chest X-rays looked better but ultimately lung function was not improved in these patients.
Beyond prednisone, many anti-inflammatory agents have been tried in this indication, summarized below:
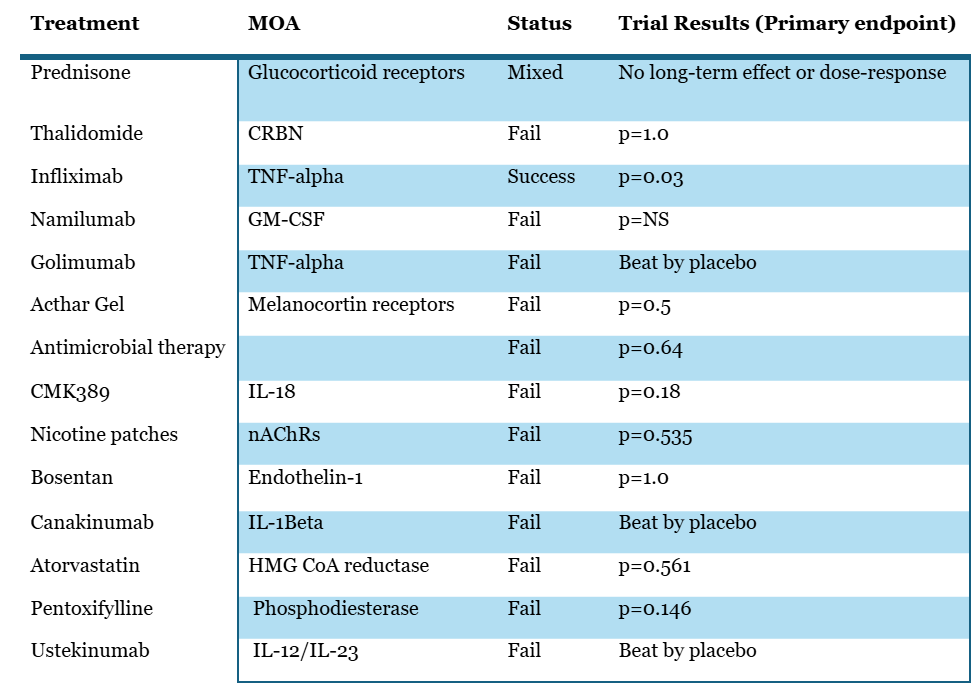
So, pretty much everything that has been tried in this indication has failed, including many of the drugs that directly lower key cytokines in the inflammatory cascade. Including biomarkers of interest to aTyr in the ph1/2 biomarker analysis such as TNFa. Once you see these results you have to ask whether inflammation is what’s driving the activity in this disease.
We also find it quite interesting that infliximab did well in its study, while golimumab (another TNFa antibody) failed its trial, quite badly. Either infliximab’s success was a random occurrence, or some differences in clinical trial design which may have led to this discordance in results.
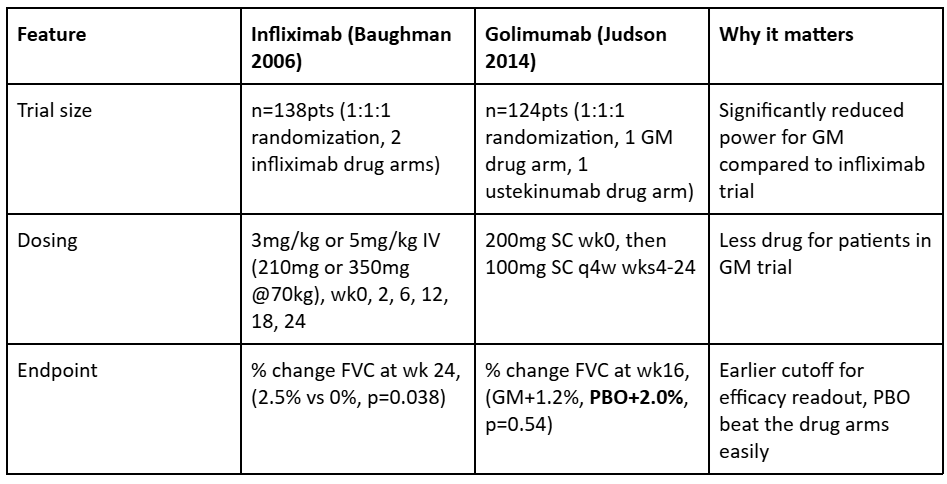
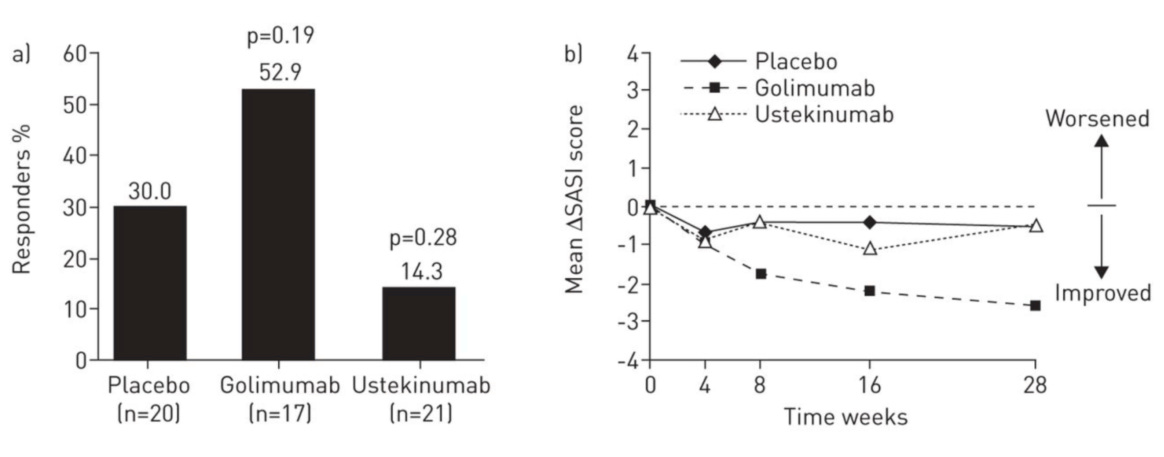
In the above table we can see that there were many clinical design features of this study which made the odds of success much lower than in the infliximab trial. Introducing a second drug in the same trial will dramatically reduce the powering of the trial. Second, lowering the dose received over time will necessarily reduce the ability for the drug to impact disease. Third, changing the endpoint to be much sooner reduces its chance of showing efficacy though we note that even at wk28 this made no difference, as placebo was ultimately the best choice for patients in this trial.
Mechanism of Action
aTyr has proposed that NRP2 agonism by efzofitimod is what will drive down inflammation and fibrosis in patients, as seen from their current slide deck.

They believe that by pushing down inflammatory signals like IL-6, TNFa, and MCP-1 that there will be a reduction in the granulomas and fibrotic tissue in patients.
They are hitching their wagon to the concept of inflammation driving disease but after the numerous failures in sarcoid by directly treating inflammation we’re wondering if they can show more to say that this molecule is resolving sarcoid specific markers or disease manifestations.
They claim that because NRP2 is upregulated in immune cells of sarcoid patients, specifically in the lung tissue, that it is overexpressed on cells to combat inflammation. The upregulation of NRP2 is in response to inflammation and in an effort to downregulate the inflammation.

While we agree it is highly expressed in these patients, we are not necessarily convinced that efzo’s binding to it necessarily leads to reduced disease. Further, I’m not convinced from the pre-clinical work that the molecule is necessarily acting as an agonist. As we’ll see in the next section, they have done some work to show it binds to NRP2 specifically, and not variants like NRP18. There is some downstream cellular response that is lowered when NRP2 is blocked. But we lack a direct experiment showing that a co-receptor phosphorylation or second-messenger changes as a result of efzofitimod binding NRP2. Even in their ligand-induced dimerization assay they never test efzofitimod alone, only in combination with other ligands. So we can’t yet be sure that this agonism is real or relevant on its own.
More troubling, when we to go back to Howard et al., 2002 investigation into HARS molecule which found the fragment 1-48 (WHEP) to be chemotactic for CCR5 bearing cells9:
The studies reported here have identified HisRS, and to a lesser extent an NH₂-terminal peptide, 1–48 HisRS, and AsnRS as aminoacyl–tRNA synthetases having proinflammatory functions.
Specifically, HisRS is a nonchemokine chemoattractant for CCR5-bearing cells that mediates its chemotactic signal by interacting with at least the third and fourth extracellular domains of the receptor, and AsnRS induces CCR3-expressing cells to migrate.
This is really problematic since CCR5 is pro-inflammatory, with evidence showing it helps to recruit and retain Th1 cells in the lung (Petrek 2002)10. We also see from the review by Russo et al., 2024 that11: In bleomycin induced fibrosis, mice deficient in both CCL3 and CCR5 exhibit reduced pulmonary influx of TGF-β1-producing cells and less fibrosis.
Even in sarcoid patients with Löfgren’s syndrome, and a specific CCR5 polymorphism, Karakaya et al, 2021 tell us that in this study12: …the expression of the chemokines in the sarcoidosis granuloma suggest that genetic variants that cause decreased or dysfunctional chemokine receptors could lead to the formation of less stable granuloma, which in turn could lead to less prolonged disease, such as the Löfgren’s syndrome phenotype of sarcoidosis.
So breaking the CCR5 receptor binding and functionality results in less granuloma, causally linking the lack of CCR5 to better patient outcomes.

We see from the Baughman 2023 paper that this CCR5 activity could directly conflict with the proposed role of efzo4.
Thus, this CCR5 element directly contradicts the proposed MoA from aTyr in two ways:
The WHEP domain has been shown to be chemotactic for CCR5, which is pro-inflammatory
The inhibition of CCR5 has been shown to result in better patient outcomes
With this in mind we have to believe the following on the MoA for it to meaningfully treat disease:
Efzo agonizes NRP2
Agonizing NRP2 is going to reduce inflammation
Reducing inflammation is going to reduce granulomas and resolve disease
The original work on WHEP domain is invalid OR CCR5 chemotaxis is not going to lead to worse disease progression
Let’s see if we can answer some of these points in the pre-clinical workup.
Pre-Clinical work on Efzo
What led the aTyr team to think they had a banger in this NRP2 binding molecule?
As we established earlier, efzofitimod lit up the Retrogenix assay for NRP2 and that made them interested in this particular target (Nangle 2025)8. Work was done to associate this target with ILD, hence the other indication being pursued. Along the path, they found out that NRP2 is highly expressed in sarcoid patients' lung tissue (specifically macrophages), naturally making it a good target in this disease. This led them to characterize the molecule with typical protein engineering techniques like SPR to characterize affinity, domain swaps on cells overexpressing the mutant NRP2 to ensure its specific to wild type NRP2 and not going to hit the similar sequence of NRP1. Ensuring it doesn’t cross react to the same location on NRP2 as other natural ligands VEGF-C and SEMA3F. They showed it has a pretty good binding, with a 23.5nM EC50 and it stays in the body long enough with a 9-11d half-life which could make monthly dosing work13.
From this perspective the results are pretty good. This is a good premise for a drug, enough to warrant taking to cell and mouse models. In there, what do we see?
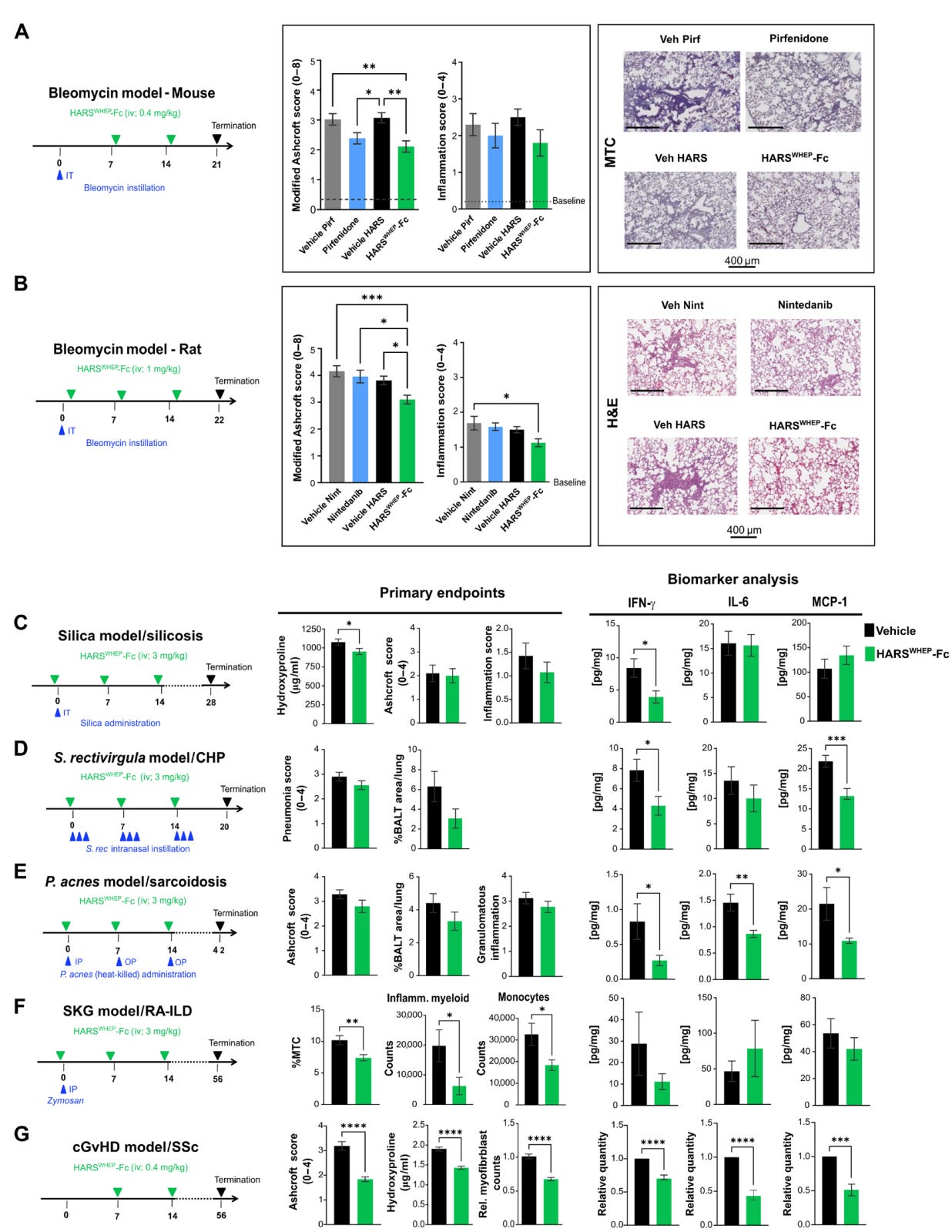
They use mouse models to try and recapitulate inflamed lung diseases, and granulomatous insult. These models have their limitations but we can at least look through them and see what happens to the granulomas and biomarkers. First, we will focus on this one, which is specifically for sarcoid:

So what’s happening here? We note that the granuloma inflammation wasn't really affected by treatment, in any model, but the biomarkers went down. This could imply that the treatment wouldn’t reverse disease in-vivo but might stop progression if you buy into the biomarkers. However, in a pre-clinical model you want to see some real activity before you feel confident about moving into patients. Also, in the above bleomycin model we see modest reductions in the composite scores for disease compared to control drug Nintedanib which is not yet proven to be effective in sarcoid patients.
In this paper they focus on the inflammation pathway and look for biomarkers of that path like IFNg, IL-6, and MCP-1. That’s fine, but keep in mind that these markers are not necessarily associated with sarcoid like, say, SAA, ACE, IL2Ra which are also investigated in their ph1/2.
Ph1/2 Clinical Data (NCT03824392)
Speaking of the Ph1/2- let’s look at the results of that study since it is the only clinical data we have to base our analysis on ahead of the ph3 readout coming in late August-Early September. This was a n=37, multi-center study in the US testing placebo and 3 drug arms to evaluate safety and efficacy. They tested 1mg/kg, 3mg/kg and 5mg/kg. They were primarily evaluating safety, but also looked at the time adjusted area under the curve of the oral corticosteroid usage (OCS) and the number of patients who maintained a taper to 5mg/day of prednisone. The idea was to taper patients off their OCS use, then see how well they could stay off of prednisone 14.
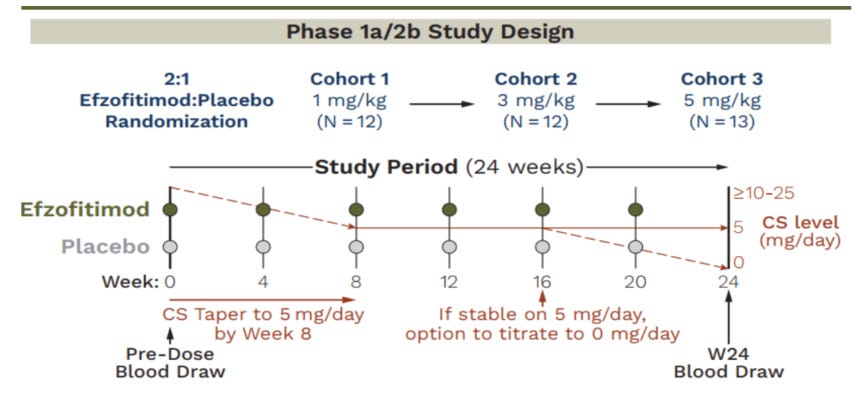

We see that they initially started with 37 patients, ITT, and then lost 9 patients mostly due to covid (n=6).
Looking at the baseline conditions we see that there is some substantial variance here:

These markers for disease burden via lung function at baseline are definitely skewing more favorably for the 3mg/kg and 5mg/kg groups. This is problematic because a baseline imbalance can confound the final results, especially in a disease where many patients will naturally resolve disease without treatment as noted above. We will also add that the FVC% variance is very high in the 5mg/kg cohort, which from our perspective suggests that there are some patients much lower and some possibly much higher than the 83.8% mean. If patients are well above the mean, it would seem almost likely that they taper off steroids and experience natural disease resolution given what we saw from Soto-Gomez, 2016.
Beyond those functional baselines and looking at the sarcoid specific biomarkers we also see this imbalance, confirming the above observations.

We’ll circle back to this later.
At the end of the trial how did the patients do? Did they taper steroids? Did they have a functional benefit in terms of lung function?
As far as steroid use goes- we would say they did not. They re-interpreted the clinical trial results from clinicaltrials.gov post-hoc to show the following table:

Suggesting a stronger change from baseline in the higher drug cohorts.
But even so, we wouldn’t call this a dose dependent response since 3mg/kg is almost the same as placebo and well within standard deviation. If we consider the pre-specified results posted to clinicaltrials.gov:
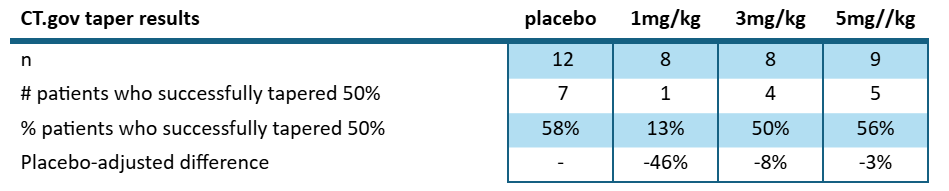
Looking at the number of patients who successfully tapered and maintained it, we see that no cohort beat placebo. That is vital to acknowledge, as this was the endpoint pre-specified by the company ahead of the ph1/2, not the post-hoc re-interpretation they did of the results featured in the Chest paper.
Looking at the amount of steroid reduction per day over the course of the study posted to clinicaltrials.gov we see:
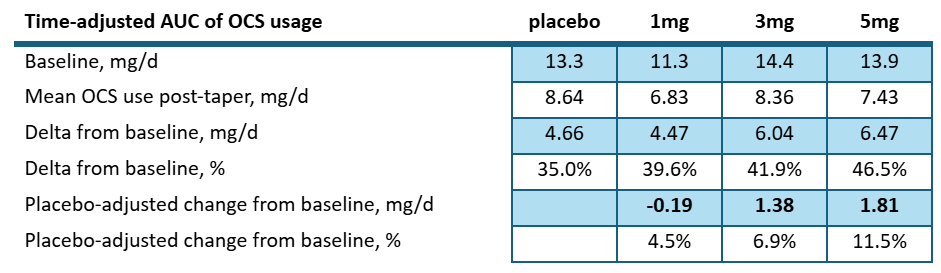
The high dose patients saw a net reduction of 1.8mg/d compared to placebo. Let’s keep that number in mind as we look into the ph3 study. But let’s also consider the most recent UpToDate article again6:

As the author notes that a small number of patients require long-term or indefinite maintenance therapy to control the disease. Is 1-3mg/d of OCS sparing truly clinically meaningful?
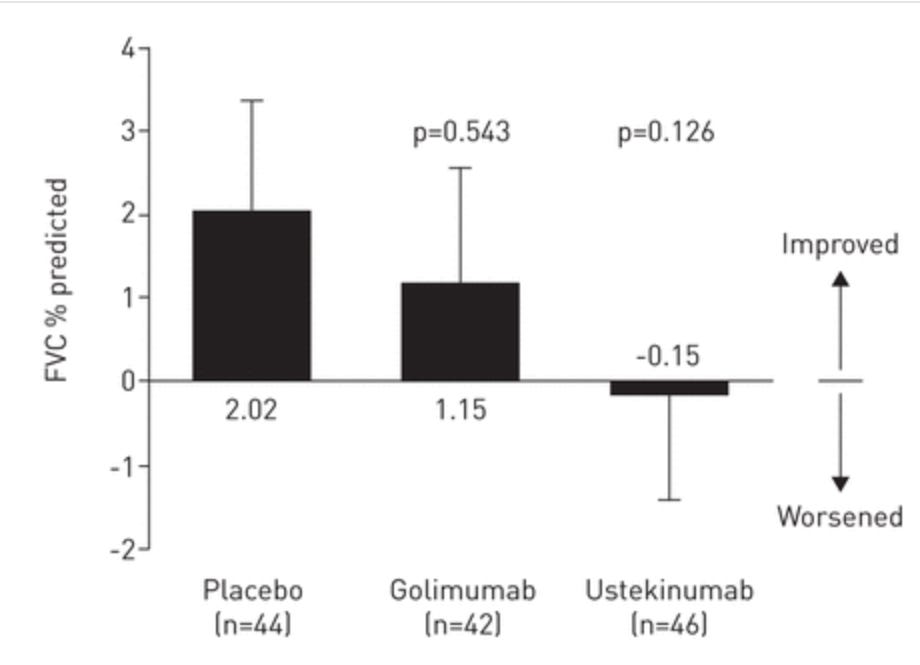
Moving on, what about the functional lung endpoint of the study, FVC?

They claim to show a dose dependent change here again. We think this shows that healthier patients regressed less.
As a comparative, let’s look at how infliximab did in a patient cohort which was substantively worse at baseline with average FVC below 70% and where half the patients were on prednisone15. In this ph2 RCT on 138 sarcoidosis patients from 34 centers across the US and Europe received either placebo, 3mg/kg or 5mg/kg of infliximab at wk0, 3, 6, 12, 18, and 24 and were then followed through to wk52. Patients who were already on prednisone and did not taper for this study, which is a key difference between this study and the efzo trial. That said, what about baselines?

We see that all the patients in this study are basically on par with the 1mg/kg group from the efzo trial, with FVC < 70%, and ACE was already reduced in these patients likely due to prednisone. Now what about the effect on lung function?
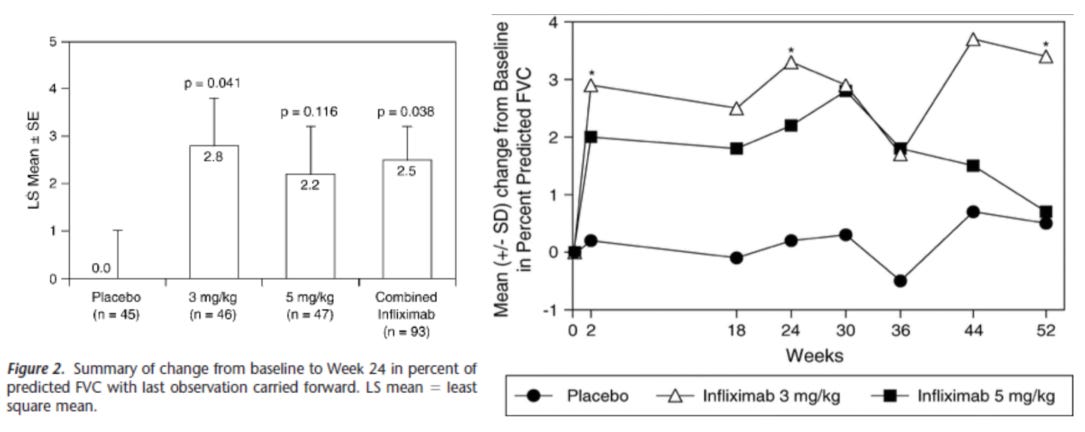
We see meaningful drug vs placebo separation, and results that clearly show infliximab is having an effect on a real functional endpoint, FVC. Further, let’s look at the biomarker data:

Keep in mind again that this patient population is entirely as sick as the 1mg/kg arm of the efzo trial, based on the FVC%. Despite this, we see that the infliximab treatment has a substantial effect on the patients compared to placebo and lowers the baseline ACE enzyme activity in the drug arms. This is important because ACE enzyme activity is highly predictive of disease burden as we’ll see in the next sections. Ultimately infliximab was able to gain off-label use in this indication, albeit as a later line therapy but this also shows the bar that a drug needs to hit in order to be considered useful to clinicians even with the strong unmet medical need of this indication.
Scroll back up and compare the FVC graphs of efzofitimod with infliximab and see if you think this drug is having a large effect on the patient cohort. We would say the efzofitimod graph is showing the difference between healthier patients at baseline more than a drug meaningfully addressing the core of the disease. We aim to prove this in the following section, but before we do that let’s sum this ph1/2 up:
Small sample size, likely confounded at baseline
Very modest effects in functional endpoint, FVC
Very modest effect in reducing use of steroids compared to placebo
Significantly worse functional improvement compared to infliximab therapy
That said, is FVC the be all and end all? No, as we know patients really don’t like being on steroids since they make patients feel worse, lead to obesity, diabetes and have effects on key organs. So, taking patients off steroids and improving or at least maintaining disease burden may be enough to warrant an approval, especially if there are few trade offs with AEs. So given the lack of functional activity from pre-clinical to clinical, we can see why aTyr decided to shift the endpoint for their phase 3 to:
Change from baseline in mean daily oral corticosteroid (OCS) dose at Week 48
This was a recent change to the endpoint that the FDA agreed on in the middle 2025 and it means that they will be considering the change in OCS use for the last month leading into the last readout, not the accrued daily OCS usage post-taper which is what they measured in the ph1/2. So, if on the final follow-up the patients are off steroids, it counts as being off steroids.
With that ph1/2 clinical data in mind, is there a way to get more certainty on whether the beneficial effects noted in the high dose arm were in fact the result of baseline imbalances or the drug effect?
To answer that, we investigated the biomarker literature to see if it was of value and ultimately predictive of disease resolution in these patients.
The Biomarkers
After reading the infliximab paper we went digging through some of the old literature from the 1980s, since Dr. Baughman references the ACE enzyme activity levels and serum ACE (sACE) in his infliximab paper. Deremee et al. 1980 found that the ACE enzyme activity level was potentially predictive of disease activity16.

But since then, we’ve learned a lot about these biomarkers. The results can have interference from ACE inhibitor drugs, different labs were better or worse at running the assay and you can have some patients who produce a genetically different version of ACE that confounds the results. It could also be confounded by other diseases like lymphoma. So, the field moved towards serum ACE (sACE) instead, but we’ll say that if you have a centralized lab working on a patient population without ACE inhibitors that don’t have the different ACE, this assay is probably pretty good as (Sunaga et al. 2022) show us with perfect specificity and PPVs of 100%17.

Beyond ACE, other biomarkers have become quite interesting in this space, which were included in the efzofitimod paper; namely IL2Ra and SAA.
The Italian group, (Zoppa et al. 2024) did a great comparative analysis on the serum biomarkers for disease in sarcoid, yielding this table below18:

With this review we can assess the value of each individual biomarker in predicting disease outcomes as per the definition of sensitivity and specificity:

We know that ACE has a high specificity, meaning it has very few false positives. We know that SAA and IL2R have high sensitivity, with low false negatives. This means that these assays are complimentary and we could combine their predictive values by using a Bayesian “rule out/ rule in” model.
We used the following values for specificity and sensitivity:
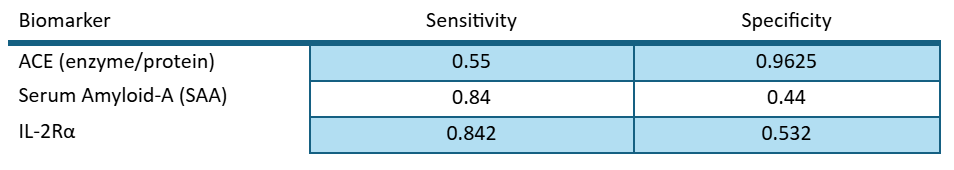
And the following algorithm for predicting whether the patients were positive or negative:

To use this model on a per patient basis, we had to also come up with patient level data. We used the ATS poster below from the ph1/2 study with the nominal baselines they established from healthy patients to prompt ChatGPT o3 to deduce and simulate a patient level data set that complies with the parameters they give:
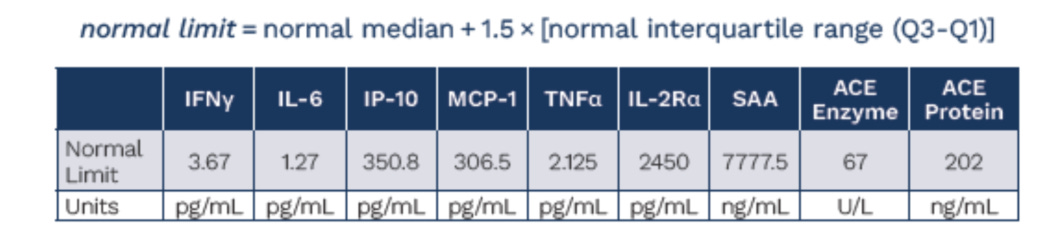

In the simulated patient level data set we have the inferred fold changes from the box plots, the same medians, the same median increase and the same % of patients in the %WNL. This ensures that we can be as close as possible to the same base data set the company has in hand but hasn’t shared publicly.
It was also noted in the poster, that patients on ACE inhibitors were removed from the ACE analysis post-hoc, and these analyses were done in a centralized lab so we don’t have to worry about that confounding19.
With all of this data we could run the Bayesian model and get essentially a probability of disease based on the biomarker data they report. From there though, we have to account for baseline imbalances because it is quite clear that the 5mg/kg group is healthier than the rest. We used FVC% to normalize the data, as a functional endpoint of disease. This yielded the following graph:
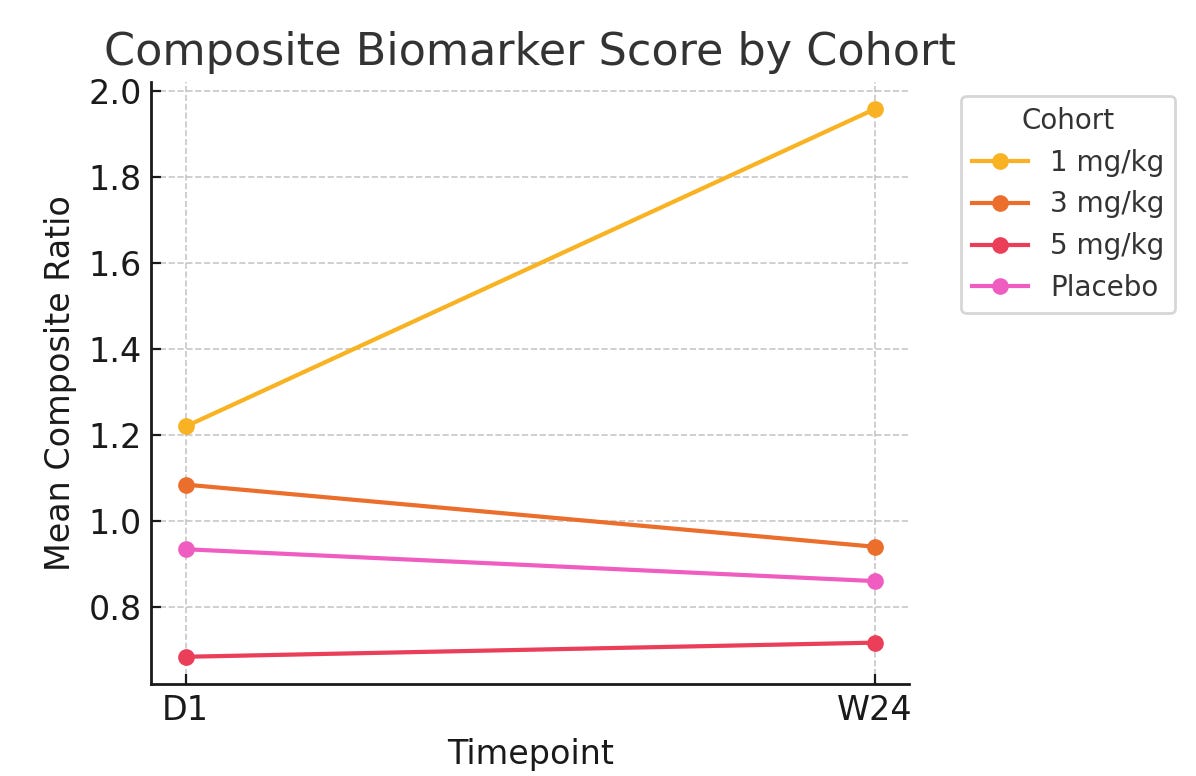
What is this showing? It’s showing what the probability of disease is, when looking at these biomarkers specifically. Line go up = bad. Line go down = better. So this graph shows that in the resulting biomarker score, patients are getting worse fast in the 1mg/kg cohort, and slowly worse in the 5mg/kg cohort (slope going up) while the placebo and 3mg/kg group are getting slightly better.
Going further, we wanted to push the analysis towards a more robust statistical approach by normalizing at the end with an ANCOVA approach (which is endorsed by the FDA) and minimize variance. In this approach we took the individual patient level data and counted the % change from baseline compared to WNL, instead of using the Bayesian approach.
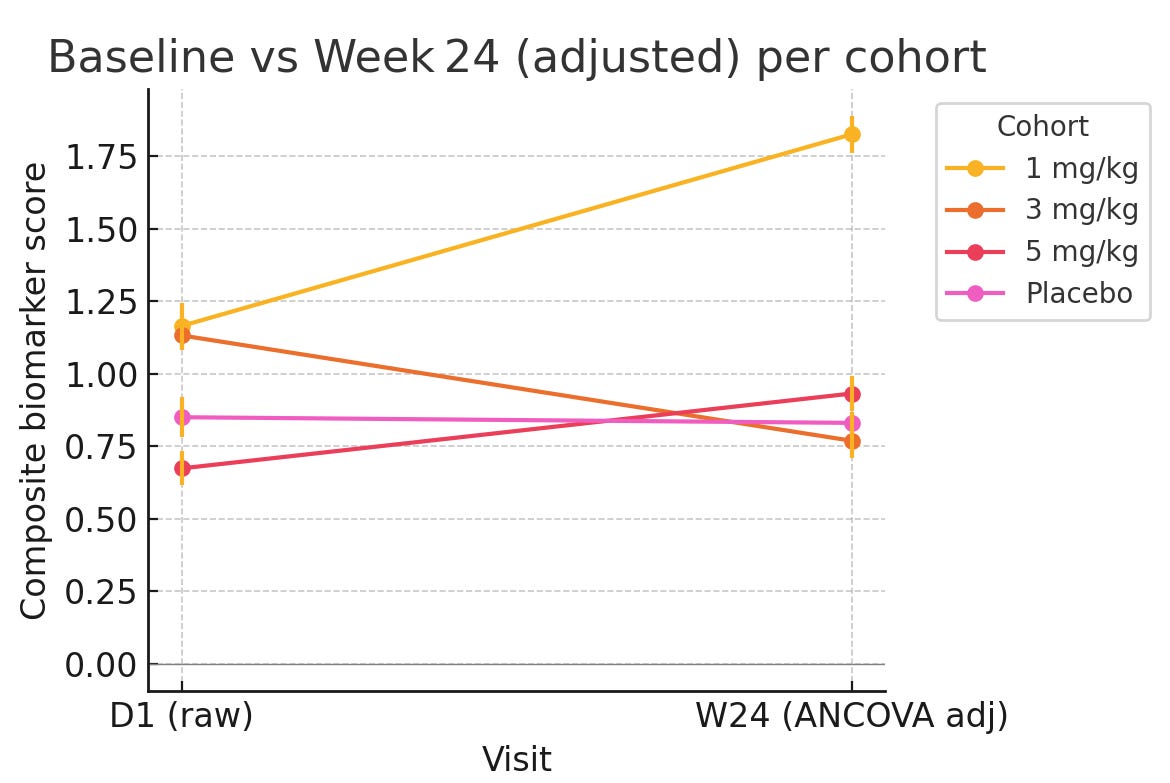
Again, we see the placebo arm beating the 5mg/kg arm here and 3mg/kg is within variance.
Is there a lot of noise looking at this very small sample set? Yes. But given these results is it worth taking a hard look at the original data and asking whether the ph1/2 is testing drug vs placebo or sick vs sicker patients all on placebo? Also yes.
Despite the very low sample size, we would argue that this makes it highly unlikely that the result observed is a result of the drug as opposed to being a chance finding given the obvious baseline imbalances.
Beyond this analysis, we want to also highlight a finding from a recent paper by Obi et al. 2025 where they re-analyze the efzofitimod ph1/2 data this time by pooling the pbo+1mg/kg group and the 3mg/kg+5mg/kg group20.
Pooling data, particularly highly imbalanced baseline data is pretty problematic. In our opinion this study highlights the results of a baseline imbalanced cohort rather than the effects of sub-therapeutic vs so-called therapeutic dosing of the drug. We see this idea exemplified by the plotting of FVC over time in the cohorts which were done using a Slopes Analysis.
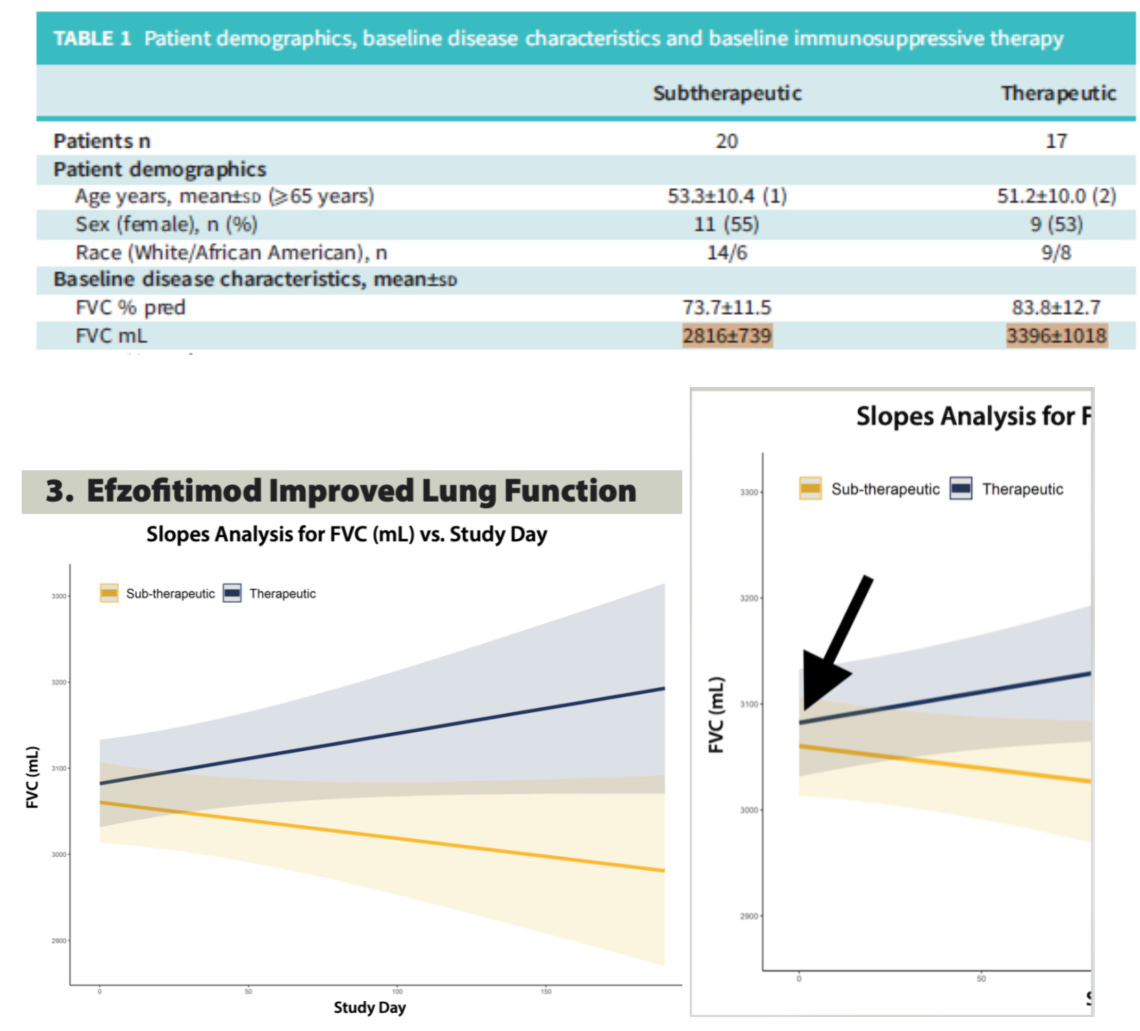
In our opinion this is a poor choice of statistical approach as it will pull these pooled cohorts closer to the overall mean at baseline via regression despite the obvious baseline differences noted in Table 1. The therapeutic cohort gets pulled down, and the sub-therapeutic cohort gets pulled up, masking the baseline imbalance. Given such an imbalance, it’s unwise to use a regression mean based analysis as a graphic here, we’d rather see a delta to baseline and a non-pooled analysis given how much worse the 1mg/kg cohort is than all the rest. However that data was already shown above and as we noted, it did not show compelling clinical activity for the drug arms compared to placebo when baseline is accounted for.
Ph3 trial and readout NCT05415137
Now that we’ve looked critically at the history of the company, the pre-clinical workup, and the ph1/2 we can look at the ph3 plan and what needs to be hit for this trial to reach statistical significance and potentially gain approval.
This is a n=268 RCT with placebo, 3mg/kg, and 5mg/kg dose arms. It is a multi-national trial with sites in the US, Europe, Brazil and Japan. Critically the endpoint shifted in this trial as mentioned previously from total OCS use over the trial to just the OCS use the last 4 weeks leading into wk48. They will also look at KSQ lung score, steroid withdrawal rate and change from baseline in absolute FVC at wk48.

As far as the baseline of these patients goes, we know from the unblinded data shown at the 2025 ATS annual meeting that initial prednisone use is lower going into the trial and that these patients are healthier at baseline than patients from the ph1/2 based on MRC dyspnea scores.

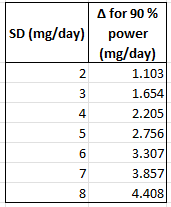
The critical “make-or-break” for this study will be whether they hit stat sig on the primary endpoint. From listening to the recent Jeffries meeting call, we heard CEO Sanjay Shukla say that they will be testing for 5mg/kg OR 3mg/kg. This is very important because it means that they will have to spend some statistical alpha in testing both dose arms for statistical significance. They will either do this with a simple Bonferroni split so either arm would have to hit p<0.025, or, they could do a Holm step-down which is similar but would require the 5mg/kg arm to hit p<0.025 and if it does, they can re-evaluate the 3mg/kg arm at p<0.05.But we think all eyes are on the 5mg/kg arm. If it hits p<0.025, they can call it a victory but our money’s on a trial failure. It is also possible they do a Hochberg, but both arms will have to hit 0.05, otherwise they will have to hit 0.025, and we still see both outcomes as highly unlikely.
We calculated the range of outcomes for the final readout. They’ll likely need to hit ~2.0-3.0mg/d delta to baseline placebo adjusted to hit stat sig. This is higher than what they saw in the ph1/2 -1.8mg/d pbo adjusted. However, with a milder patient population and the baseline imbalances being corrected we think they miss stat sig and most patients come off prednisone entirely.
In summary
Company is based on leveraging unique knowledge in tRNA synthetases which have failed to find success in the clinic
Novel target biology springs hope, but conflicting activities in the MoA invite more questions than answers
Pre-clinical data fails to demonstrate clear, translatable activity in disease specific models
Ph1/2 data is confounded by baseline imbalances across multiple markers of disease
Statistical analyses can remove the drug specific effect
Ph3 requires substantive change in OCS use compared to baseline on a placebo adjusted basis, which seems unlikely given the above points
References
1. Suzuki M, Kato C, Kato A. Therapeutic antibodies: their mechanisms of action and the pathological findings they induce in toxicity studies. J Toxicol Pathol. 2015;28(3):133-139. doi:10.1293/tox.2015-0031
2. Zhou JJ, Wang F, Xu Z, et al. Secreted Histidyl-tRNA Synthetase Splice Variants Elaborate Major Epitopes for Autoantibodies in Inflammatory Myositis. J Biol Chem. 2014;289(28):19269-19275. doi:10.1074/jbc.C114.571026
3. Lo WS, Gardiner E, Xu Z, et al. Human tRNA Synthetase Catalytic Nulls with Diverse Functions. Science. 2014;345(6194):328-332. doi:10.1126/science.1252943
4. Baughman R, Culver D. Efzofitimod: a novel anti-inflammatory agent for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2023;40(1):e2023002. doi:10.36141/svdld.v40i1.13617
5. Soto-Gomez N, Peters JI, Nambiar AM. Diagnosis and Management of Sarcoidosis. Am Fam Physician. 2016;93(10):840-850.
6. Culver D, Flaherty K, Dieffenbach P. Treatment of pulmonary sarcoidosis: Initial approach. UpToDate. Published online November 2024.
7. Paramothayan NS, Lasserson TJ, Jones P. Corticosteroids for pulmonary sarcoidosis. Cochrane Airways Group, ed. Cochrane Database Syst Rev. 2005;2010(5). doi:10.1002/14651858.cd001114.pub2
8. Nangle LA, Xu Z, Siefker D, et al. A human histidyl-tRNA synthetase splice variant therapeutic targets NRP2 to resolve lung inflammation and fibrosis. Sci Transl Med. 2025;17(789). doi:10.1126/scitranslmed.adp4754
9. Howard OMZ, Dong HF, Yang D, et al. Histidyl–tRNA Synthetase and Asparaginyl–tRNA Synthetase, Autoantigens in Myositis, Activate Chemokine Receptors on T Lymphocytes and Immature Dendritic Cells. J Exp Med. 2002;196(6):781-791. doi:10.1084/jem.20020186
10. Petrek M, Gibejova A, Drabek J, et al. CC chemokine receptor 5 (CCR5) mRNA expression in pulmonary sarcoidosis. Immunol Lett. 2002;80(3):189-193. doi:10.1016/S0165-2478(01)00324-8
11. Russo RC, Ryffel B. The Chemokine System as a Key Regulator of Pulmonary Fibrosis: Converging Pathways in Human Idiopathic Pulmonary Fibrosis (IPF) and the Bleomycin-Induced Lung Fibrosis Model in Mice. Cells. 2024;13(24):2058. doi:10.3390/cells13242058
12. Karakaya B, van Moorsel CHM, Veltkamp M, et al. A Polymorphism in C-C Chemokine Receptor 5 (CCR5) Associates with Löfgren’s Syndrome and Alters Receptor Expression as well as Functional Response. Cells. 2021;10(8):1967. doi:10.3390/cells10081967
13. Walker G, Butterfield R, Mathews K, et al. Results of a Phase 1b/2 Study of ATYR1940 in adolescents and young adults with early onset facioscapulohumeral muscular dystrophy (FSHD) (ATYR1940-C-003). Neuromuscul Disord. 2017;27:S199. doi:10.1016/j.nmd.2017.06.381
14. Culver DA, Aryal S, Barney J, et al. Efzofitimod for the Treatment of Pulmonary Sarcoidosis. Chest. 2023;163(4):881-890. doi:10.1016/j.chest.2022.10.037
15. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174(7):795-802. doi:10.1164/rccm.200603-402OC
16. DeREMEE RA, Rohrbach MS. Serum Angiotensin-Converting Enzyme Activity in Evaluating the Clinical Course of Sarcoidosis. Ann Intern Med. 1980;92(3):361-365. doi:10.7326/0003-4819-92-3-361
17. Sunaga N, Koga Y, Hachisu Y, et al. Role of Neuron-Specific Enolase in the Diagnosis and Disease Monitoring of Sarcoidosis. Turner A, ed. Can Respir J. 2022;2022:1-9. doi:10.1155/2022/3726395
18. Della Zoppa M, Bertuccio FR, Campo I, et al. Phenotypes and Serum Biomarkers in Sarcoidosis. Diagnostics. 2024;14(7):709. doi:10.3390/diagnostics14070709
20. Obi ON, Baughman RP, Crouser ED, et al. Therapeutic doses of efzofitimod demonstrate efficacy in pulmonary sarcoidosis. ERJ Open Res. 2025;11(1). doi:10.1183/23120541.00536-2024
If only I had discovered this and sold at the time... Thank you very much for your detailed work!